
Thailand
Kingdom of Thailand
| |
|---|---|
| Anthem: เพลงชาติไทย Phleng Chat Thai "Thai National Anthem" | |
.svg)  | |
| Capital and largest city | Bangkok[a] 13°48′N 100°33′E / 13.800°N 100.550°E |
| Official languages | Thai[1] |
Spoken languages | Central Thai, Isan, Lanna (Northern Thai), Dambro (Southern Thai), Karen, Pattani Malay, Bangkok Malay, Teochew, Hokkien |
| Ethnic groups |
|
| Religion (2018 census)[2] |
|
| Demonym(s) | Thai |
| Government | Unitary parliamentary constitutional monarchy |
• Monarch | Vajiralongkorn (Rama X) |
| Paetongtarn Shinawatra | |
| Legislature | National Assembly |
| Senate | |
| House of Representatives | |
| Formation | |
| 1238–1438 | |
| 1351–1767 | |
| 1767–1782 | |
| 6 April 1782 | |
| 24 June 1932 | |
| 6 April 2017 | |
| Area | |
• Total | 513,120 km2 (198,120 sq mi) (50th) |
• Water (%) | 0.4 (2,230 km2) |
| Population | |
• 2024 estimate | |
• 2010 census | 64,785,909[4] (21st) |
• Density | 132.1/km2 (342.1/sq mi) (88th) |
| GDP (PPP) | 2024 estimate |
• Total | |
• Per capita | |
| GDP (nominal) | 2024 estimate |
• Total | |
• Per capita | |
| Gini (2021) | medium inequality |
| HDI (2022) | very high (66th) |
| Currency | Thai baht (฿) (THB) |
| Time zone | UTC+7 (ICT) |
| Date format | dd/mm/yyyy (BE) |
| Drives on | left |
| Calling code | +66 |
| ISO 3166 code | TH |
| Internet TLD | |
| |


Thailand,[i] officially the Kingdom of Thailand and historically known as Siam (the official name until 1939),[ii] is a country in Southeast Asia on the Indochinese Peninsula. With a population of almost 66 million,[8] it spans 513,115 square kilometres (198,115 sq mi).[9] Thailand is bordered to the northwest by Myanmar, to the northeast and east by Laos, to the southeast by Cambodia, to the south by the Gulf of Thailand and Malaysia, and to the southwest by the Andaman Sea; it also shares maritime borders with Vietnam to the southeast and Indonesia and India to the southwest. Bangkok is the state capital and largest city.[10]
Thai peoples migrated from southwestern China to mainland Southeast Asia from the 6th to 11th centuries. Indianised kingdoms such as the Mon, Khmer Empire, and Malay states ruled the region, competing with Thai states such as the Kingdoms of Ngoenyang, Sukhothai, Lan Na, and Ayutthaya, which also rivalled each other. European contact began in 1511 with a Portuguese diplomatic mission to Ayutthaya, which became a regional power by the end of the 15th century. Ayutthaya reached its peak during the 18th century, until it was destroyed in the Burmese–Siamese War. King Taksin the Great quickly reunified the fragmented territory and established the short-lived Thonburi Kingdom (1767–1782), of which he was the only king. He was succeeded in 1782 by Phutthayotfa Chulalok (Rama I), the first monarch of the current Chakri dynasty. Throughout the era of Western imperialism in Asia, Siam remained the only state in the region to avoid colonization by foreign powers, although it was often forced to make territorial, trade, and legal concessions in unequal treaties.[11] The Siamese system of government was centralised and transformed into a modern unitary absolute monarchy during the 1868–1910 reign of Chulalongkorn (Rama V). In World War I, Siam sided with the Allies, a political decision made in order to amend the unequal treaties. Following a bloodless revolution in 1932, it became a constitutional monarchy and changed its official name to Thailand, becoming an ally of Japan in World War II. In the late 1950s, a military coup under Sarit Thanarat revived the monarchy's historically influential role in politics. During the Cold War, Thailand became a major ally of the United States and played an anti-communist role in the region as a member of SEATO, which was disbanded in 1977.
Apart from a brief period of parliamentary democracy in the mid-1970s and 1990s, Thailand has periodically alternated between democracy and military rule. Since the 2000s, the country has been in continual political conflict between supporters and opponents of twice-elected Prime Minister of Thailand Thaksin Shinawatra, which resulted in two coups (in 2006 and 2014), along with the establishment of its current constitution, a nominally democratic government after the 2019 Thai general election, and large pro-democracy protests in 2020–2021, which included unprecedented demands to reform the monarchy. Since 2019, it has been nominally a parliamentary constitutional monarchy; in practice, however, structural advantages in the constitution have ensured the military's continued influence in politics.[12]
Thailand is a middle power in global affairs and a founding member of ASEAN. It has the second-largest economy in Southeast Asia and the 23rd-largest in the world by PPP, and it ranks 91st by nominal GDP per capita. Thailand is classified as a newly industrialised economy, with manufacturing, agriculture, and tourism as leading sectors.[13][14]
Etymology
Thailand[i] was known by outsiders prior to 1939 as Siam.[ii] According to George Cœdès, the word Thai (ไทย) means 'free man' in the Thai language, "differentiating the Thai from the natives encompassed in Thai society as serfs".[15]: 197 According to Chit Phumisak, Thai (ไท) simply means 'people' or 'human being'; his investigation shows that some rural areas used the word "Thai" instead of the usual Thai word khon (คน) for people.[16] According to Michel Ferlus, the ethnonyms Thai-Tai (or Thay-Tay) would have evolved from the etymon *k(ə)ri: 'human being'.[iii][18]
Thais often refer to their country using the polite form prathet Thai (Thai: ประเทศไทย). They also use the more colloquial term mueang Thai (Thai: เมืองไทย) or simply Thai; the word mueang, archaically referring to a city-state, is commonly used to refer to a city or town as the centre of a region. Ratcha Anachak Thai (Thai: ราชอาณาจักรไทย) means 'kingdom of Thailand' or 'kingdom of Thai'. Etymologically, its components are: ratcha (Sanskrit: राजन्, rājan, 'king, royal, realm'), ana- (Pali āṇā 'authority, command, power', itself from the Sanskrit आज्ञा, ājñā, of the same meaning), and -chak (from Sanskrit चक्र cakra- 'wheel', a symbol of power and rule). The Thai National Anthem (Thai: เพลงชาติ), written by Luang Saranupraphan during the patriotic 1930s, refers to the Thai nation as prathet Thai (Thai: ประเทศไทย). The first line of the national anthem is: prathet thai ruam lueat nuea chat chuea thai (Thai: ประเทศไทยรวมเลือดเนื้อชาติเชื้อไทย), 'Thailand is founded on blood and flesh'.[19]
The former name Siam may have originated from Sanskrit श्याम (śyāma, 'dark')[16] or Mon ရာမည (rhmañña, 'stranger'), probably the same root as Shan and Assam.[20] The word Śyâma is possibly not the true origin, but a pre-designed deviation from its proper, original meaning.[21][22] Another theory is the name derives from the Chinese calling this region 'Xian'.[iv][23]: 8 The ancient Khmers used the word Siam to refer to people settled in the west Chao Phraya River valley surrounding the ancient city of Nakhon Pathom in the present-day central Thailand; it may probably originate from the name of Lord Krishna, which also called Shyam, as in the Wat Sri Chum Inscription, dated 13th century CE, mentions Phra Maha Thera Sri Sattha came to restore Phra Pathommachedi at the city of Lord Krishna (Nakhon Pathom) in the early era of the Sukhothai Kingdom.[24]
_of_Siam_Signature_(English).svg)
The signature of King Mongkut (r. 1851–1868) reads SPPM (Somdet Phra Poramenthra Maha) Mongkut Rex Siamensium (Mongkut, King of the Siamese). This usage of the name in the country's first international treaty gave the name Siam official status, until 24 June 1939 when it was changed to Thailand.[25]
History
This section may need to be rewritten to comply with Wikipedia's quality standards, as many reasons, see talk page. (February 2024) |
Prehistory and origins
There is evidence of continuous human habitation in present-day Thailand from 20,000 years ago to the present day.[26]: 4 The earliest evidence of rice growing is dated at 2,000 BCE.[27]: 4 Areas comprising what is now Thailand participated in the Maritime Jade Road, as ascertained by archeological research. The trading network existed for 3,000 years, between 2000 BCE to 1000 CE.[28][29][30][31] Bronze appeared c. 1,250–1,000 BCE.[27]: 4 The site of Ban Chiang in northeast Thailand currently ranks as the earliest known centre of copper and bronze production in Southeast Asia.[32] Iron appeared around 500 BCE.[27]: 5 The Kingdom of Funan was the first and most powerful Southeast Asian kingdom at the time (2nd century BCE).[26]: 5 The Mon people established the principalities of Dvaravati and Kingdom of Hariphunchai in the 6th century. The Khmer people established the Khmer empire, centred in Angkor, in the 9th century.[26]: 7 Tambralinga, a Malay state controlling trade through the Malacca Strait, rose in the 10th century.[26]: 5 The Indochina peninsula was heavily influenced by the culture and religions of India from the time of the Kingdom of Funan to that of the Khmer Empire.[33]
The Thai people are of the Tai ethnic group, characterized by common linguistic roots.[34]: 2 Chinese chronicles first mention the Tai peoples in the 6th century BCE. While there are many assumptions regarding the origin of Tai peoples, David K. Wyatt, a historian of Thailand, argued that their ancestors who at present inhabit Laos, Thailand, Myanmar, India, and China came from the Điện Biên Phủ area between the 5th and the 8th century.[34]: 6 Thai people began migrating into present-day Thailand gradually from the 6th to 11th century, which Mon and Khmer people occupied at the time.[35] Thus Thai culture was influenced by Indian, Mon, and Khmer cultures.[36]: 203 Tai people intermixed with various ethnic and cultural groups in the region, resulting in many groups of present-day Thai people.[v] Genetic evidences suggested that ethnolinguistics could not accurately predict the origins of the Thais.[37][38][39] Sujit Wongthes argued that Thai is not a race or ethnicity but a culture group.[40]
According to French historian George Cœdès, "The Thai first enter history of Farther India in the eleventh century with the mention of Syam slaves or prisoners of war in Champa epigraphy", and "in the twelfth century, the bas-reliefs of Angkor Wat" where "a group of warriors" are described as Syam,[15]: 190–191, 194–195 though Cham accounts do not indicate the origins of Syam or what ethnic group they belonged to.[41] The origins and ethnicity of the Syam remain unclear, with some literature suggesting that Syam refers to the Shan people, the Bru people, or the Brau people.[41][42] However, mainland Southeast Asian sources from before the fourteenth century primarily used the word Syam as an ethnonym, referring to those who belonged to a separate cultural category different from the Khmer, Cham, Bagan, or Mon. This contrasts with the Chinese sources, where Xian was used as a toponym.[41]
Early Tai confederate cities: (691 BCE – 13th century CE)

.png)
Theoretically, Tai-Kadai-speaking people formed as early as the 12th century BCE in the middle of the Yangtze basin. Some groups later migrated south to Guangxi.[43] However, after several bloody centuries against Chinese influence in Guangxi from the 333 BCE-11th centuries, hundreds of thousands of Tais were killed,[44]: 5 [45]: 193, 239–249 thus, Tai people began to move southwestward along the rivers and over the lower passes into the mountain north of Southeast Asia and river valleys in present-day Assam of India.[46][47] Some evidence indicates that the ancestors of Tai people migrated en masse southwestwards out of Yunnan only after the 1253 Mongol invasion of Dali, but not generally accepted.[48]: 38
Tais defeated indigenous tribes and emerged as the new power in the new region, several Tai city-states were established, scattered from Điện Biên Phủ in present-day northwestern Vietnam and highland Southeast Asia to northeastern India.[49][50][51] According to the Simhanavati legend given in several chronicles, the first Tai city-state in northern Thailand, Singhanavati, was found around the 7th century;[52]: 5, 9 however, several modern geology and archaeology studies found that its center, Yonok Nahaphan, dates from 691 BCE–545 CE,[52]: 7 [53][54][55] coinciding roughly with the establishment of Shan States, another Tai's federated principalities in the present-day northeast Myanmar.[56][57][58][59] as well as Muang Sua (Luang Prabang) in the east.[60][61] After Singhanavati was submerged below Chiang Saen Lake due to an earthquake in 545,[53][55][62] the survivors then founded a new seat at Wieng–Prueksha, the kingdom lasted for another 93 years.[63]
In addition to Singhanavati, another northern principality probably related to the Tai people, Ngoenyang, was established as the successor of Singhanavati in 638 by Lavachakkaraj, also centered in Wieng–Prueksha (present-day Mae Sai District, Chiang Rai).[23]: 8 Its seat was moved to Chiang Mai in 1262 by King Mangrai, which considered the foundation of the Lan Na kingdom. Mangrai unified the surrounding area and also created a network of states through political alliances to the east and north of the Mekong. His dynasty would rule the kingdom continuously for the next two centuries.[23]: 8 Lan Na expanded its territory southward and annexed the Mon Hariphunchai of Dvaravati in 1292.[64]: 208
In the late 10 century, Tai people began to migrate further south to the present-day upper central Thailand.[65]: 46–9, 83–6 Around the 1100s period, several cities in this area, such as Songkwae, Sawankhalok, and Chakangrao, were ruled by the Tai people, and they eventually launched several battles against the pre-existing Mon of Lavo, who had been falling under Chenla and Khmer influences since the 7th century, thus bringing the establishment of the Tai people's independent state, Sukhothai Kingdom, in the upper Chao Phraya River valley in 1238.[34]: 52–3
The earliest conflict between Tai people and the preexisting ethnics was recorded in the mid-4th century when the ruler of Singhanavati, Pangkharat, forcibly lost the seat at Yonok to King Khom from Umongasela (present-day Fang). He then fled to Vieng Si Tuang (เวียงศรีทวง; present-day Wiang Phang Kham, Mae Sai district) but had to send tributes to Yonok annually until his son, Phrom, took back Yonok and expelled King Khom from Umongasela.[66][67][68] Phrom also marched the troops south to occupy Chakangrao from the enemy as well as founding the city of Songkwae.[66] Some historians suggest that Lavo's capital, Lopburi, was once seized by Phrom.[66] In contrast, Tai people instead established relationships with Siamese Mon via royal intermarriages.[65]: 46–9, 83–6
Mon and Lavo Kingdoms: (5th century CE – 13th century CE)


As is generally known, the present-day Thai people were previously called Siamese before the country was renamed Thailand in the mid-20th century.[16] Several genetic studies published in the 21st century suggest that the so-called Siamese people (central Thai) might have had Mon origins since their genetic profiles are more closely related to the Mon people in Myanmar than the Tais in southern China,[37] and they probably later became Tais via cultural diffusion after the arriving of Tai people from the north around the 8th–10th centuries.[51][69][70] This is also reflected in the language since over half of the vocabulary in the central Thai language is derived from or borrowed from the Mon language as well as Pali and Sanskrit.[69][71] Moreover, the Jinakalamali chronicle of Tai's Lan Na also called the southern region occupied by the Mon Haripuñjaya of Dvaravati as Shyam Pradesh (lit. 'the land of Siam people'), which indicates that the ancient Siamese and the Mon people in central Thailand were probably the same ethnolinguistic group.[72]
The earliest evidence to mention the Siam people are stone inscriptions found in Angkor Borei of Funan (K.557 and K.600), dated 661 CE, the slave's name is mentioned as "Ku Sayam" meaning "Sayam female slaves" (Ku is a prefix used to refer to female slaves in the pre-Angkorian era), and the Takéo inscriptions (K.79) written in 682 during the reign of Bhavavarman II of Chenla also mention Siam Nobel: Sāraṇnoya Poña Sayam, which was transcribed into English as: the rice field that was given to the poña (noble rank) who was called Sayam (Siam).[73] The Song Huiyao Jigao (960–1279) indicate Siamese people settled in the west central Thailand and their state was called Xiān guó (Chinese: 暹國), while the eastern plain belonged to the Mon of Lavo (Chinese: 羅渦國),[74] who later fell under the Chenla and Khmer hegemony around the 7th–9th centuries.[75] Those Mon political entities, which also included Haripuñjaya in the north and several city-states in the northeast, are collectively called Dvaravati. However, the states of Siamese Mon and Lavo were later merged via the royal intermarriage and became Ayutthaya Kingdom in the mid-14th century,[74] while the southwestern Isan principalities, centered in Phanom Rung and Phimai, later pledged allegiance to Siamese's Ayutthaya during the reign of Borommarachathirat II (r. 1424–1448).[76] The remaining principal city-states in Isan region became Lan Xang around 1353 after the twin cities of Muang Sua (Luang Prabang) and Vieng Chan Vieng Kham (Vientiane) became independent following the death of the Sukhothai king Ram Khamhaeng.[77]: 51
According to the Wat Kud Tae inscription (K.1105), dated c. 7th century, during the period that the eastern Mon entity, Lavo, was strongly influenced by the Chenla, the Siamese Mon in the west also established a royal intermarriage with Chenla as Sri Chakatham, prince of Sambhuka (ศามภูกะ, in the present-day Ratchaburi province), married to a princess of Isanavarman I, and two mandalas then became an ally.[78] After Chenla sieged Funan and moved the center to Angkor, both Siamese Mon and the Angkorian eventually marched the troops to attack Vijaya of Champa in 1201 during the reign of Jayavarman VII, as recorded in the Cho-Dinh inscription (C.3).[79]
Sukhothai Kingdom (1238 CE – 14th century CE)

.jpg)

After the decline of the Khmer Empire and Kingdom of Pagan in the early 13th century, various states thrived in their place. The domains of Tai people existed from the northeast of present-day India to the north of present-day Laos and to the Malay Peninsula.[34]: 38–9 During the 13th century, Tai people had already settled in the core land of Dvaravati and Lavo Kingdom to Nakhon Si Thammarat in the south. There are, however, no records detailing the arrival of the Tais.[34]: 50–1
Around 1240, Pho Khun Bang Klang Hao, a local Tai ruler, rallied the people to rebel against the Khmer. He later crowned himself the first king of Sukhothai Kingdom in 1238.[34]: 52–3 Mainstream Thai historians count Sukhothai as the first kingdom of Thai people. Sukhothai expanded furthest during the reign of Ram Khamhaeng (r. 1279–1298). However, it was mostly a network of local lords who swore fealty to Sukhothai, not directly controlled by it.[34]: 55–6 He is believed have invented Thai script and Thai ceramics were an important export in his era. Sukhothai embraced Theravada Buddhism in the reign of Maha Thammaracha I (1347–1368).
Ayutthaya Kingdom (1351–1767)



According to the most widely accepted version of its origin, the Ayutthaya Kingdom rose from the earlier, nearby Lavo Kingdom and Suvarnabhumi with Uthong as its first king. Ayutthaya was a patchwork of self-governing principalities and tributary provinces owing allegiance to the King of Ayutthaya under the mandala system.[80]: 355 Its initial expansion was through conquest and political marriage. Before the end of the 15th century, Ayutthaya invaded the Khmer Empire three times and sacked its capital Angkor.[81]: 26 Ayutthaya then became a regional power in place of the Khmer. Constant interference of Sukhothai effectively made it a vassal state of Ayutthaya and it was finally incorporated into the kingdom. Borommatrailokkanat brought about bureaucratic reforms which lasted into the 20th century and created a system of social hierarchy called sakdina, where male commoners were conscripted as corvée labourers for six months a year.[82]: 107 Ayutthaya was interested in the Malay Peninsula, but failed to conquer the Malacca Sultanate which was supported by the Chinese Ming dynasty.[26]: 11, 13
European contact and trade started in the early-16th century, with the envoy of Portuguese duke Afonso de Albuquerque in 1511. Portugal became an ally and ceded some soldiers to King Rama Thibodi II.[83] The Portuguese were followed in the 17th century by the French, Dutch, and English. Rivalry for supremacy over Chiang Mai and the Mon people pitted Ayutthaya against the Burmese Kingdom. Several wars with its ruling Taungoo dynasty starting in the 1540s in the reign of Tabinshwehti and Bayinnaung were ultimately ended with the capture of the capital in 1570.[82]: 146–7 Then was a brief period of vassalage to Burma until Naresuan proclaimed independence in 1584.[23]: 11
Ayutthaya then sought to improve relations with European powers for many successive reigns. The kingdom especially prospered during cosmopolitan Narai's reign (1656–1688) when some European travelers regarded Ayutthaya as an Asian great power, alongside China and India.[27]: ix However, growing French influence later in his reign was met with nationalist sentiment and led eventually to the Siamese revolution of 1688.[82]: 185–6 However, overall relations remained stable, with French missionaries still active in preaching Christianity.[82]: 186
After a bloody period of dynastic struggle, Ayutthaya entered into what has been called the Siamese "golden age", a relatively peaceful episode in the second quarter of the 18th century when art, literature, and learning flourished. There were seldom foreign wars, apart from conflict with the Nguyễn lords for control of Cambodia starting around 1715. The last fifty years of the kingdom witnessed bloody succession crises, where there were purges of court officials and able generals for many consecutive reigns. In 1765, a combined 40,000-strong force of Burmese armies invaded it from the north and west.[84]: 250 The Burmese under the new Alaungpaya dynasty quickly rose to become a new local power by 1759. After a 14-month siege, the capital city's walls fell and the city was burned in April 1767.[85]: 218
Thonburi Kingdom (1767–1782)

The capital and many of its territories lay in chaos after the war. The former capital was occupied by the Burmese garrison army and five local leaders declared themselves overlords, including the lords of Sakwangburi, Phitsanulok, Pimai, Chanthaburi, and Nakhon Si Thammarat. Chao Tak, a capable military leader, proceeded to make himself a lord by right of conquest, beginning with the legendary sack of Chanthaburi. Based at Chanthaburi, Chao Tak raised troops and resources, and sent a fleet up the Chao Phraya to take the fort of Thonburi. In the same year, Chao Tak was able to retake Ayutthaya from the Burmese only seven months after the fall of the city.[86]
Chao Tak then crowned himself as Taksin and proclaimed Thonburi as temporary capital in the same year. He also quickly subdued the other warlords. His forces engaged in wars with Burma, Laos, and Cambodia, which successfully drove the Burmese out of Lan Na in 1775,[82]: 225 captured Vientiane in 1778[82]: 227–8 and tried to install a pro-Thai king in Cambodia in the 1770s. In his final years there was a coup, caused supposedly by his "insanity", and eventually Taksin and his sons were executed by his longtime companion General Chao Phraya Chakri (the future Rama I). He was the first king of the ruling Chakri dynasty and founder of the Rattanakosin Kingdom on 6 April 1782.[citation needed]
Rattanakosin Kingdom and modernization (1782 –1932)

.jpg)


Under Rama I (1782–1809), Rattanakosin successfully defended against Burmese attacks and put an end to Burmese incursions. He also created suzerainty over large portions of Laos and Cambodia.[87] In 1821, Briton John Crawfurd was sent to negotiate a new trade agreement with Siam – the first sign of an issue which was to dominate 19th century Siamese politics.[88] Bangkok signed the Burney Treaty in 1826, after the British victory in the First Anglo-Burmese War.[82]: 281 Anouvong of Vientiane, who mistakenly held the belief that Britain was about to launch an invasion of Bangkok, started the Lao rebellion in 1826 which was suppressed.[82]: 283–5 Vientiane was destroyed and a large number of Lao people were relocated to Khorat Plateau as a result.[82]: 285–6 Bangkok also waged several wars with Vietnam, where Siam successfully regained hegemony over Cambodia.[82]: 290–2
From the late-19th century, Siam tried to rule the ethnic groups in the realm as colonies.[82]: 308 In the reign of Mongkut (1851–1868), who recognised the potential threat Western powers posed to Siam, his court contacted the British government directly to defuse tensions.[82]: 311 A British mission led by Sir John Bowring, Governor of Hong Kong, led to the signing of the Bowring Treaty, the first of many unequal treaties with Western countries. This, however, brought trade and economic development to Siam.[89] The unexpected death of Mongkut from malaria led to the reign of underage King Chulalongkorn, with Somdet Chaophraya Sri Suriwongse (Chuang Bunnag) acting as regent.[82]: 327
Chulalongkorn (r. 1868–1910) initiated centralisation, set up a privy council, and abolished slavery and the corvée system. The Front Palace crisis of 1874 stalled attempts at further reforms.[82]: 331–3 In the 1870s and 1880s, he incorporated the protectorates up north into the kingdom proper, which later expanded to the protectorates in the northeast and the south.[82]: 334–5 He established twelve krom in 1888, which were equivalent to present-day ministries.[82]: 347 The crisis of 1893 erupted, caused by French demands for Laotian territory east of Mekong.[82]: 350–3 Thailand is the only Southeast Asian state never to have been colonised by a Western power,[90] in part because Britain and France agreed in 1896 to make the Chao Phraya valley a buffer state.[91] Not until the 20th century could Siam renegotiate every unequal treaty dating from the Bowring Treaty, including extraterritoriality. The advent of the monthon system marked the creation of the modern Thai nation-state.[82]: 362–3 In 1905, there were unsuccessful rebellions in the ancient Patani area, Ubon Ratchathani, and Phrae in opposition to an attempt to blunt the power of local lords.[82]: 371–3
The Palace Revolt of 1912 was a failed attempt by Western-educated military officers to overthrow the Siamese monarchy.[82]: 397 Vajiravudh (r. 1910–1925) responded by propaganda for the entirety of his reign,[82]: 402 which promoted the idea of the Thai nation.[82]: 404 In 1917, Siam joined the First World War on the side of the Allies.[82]: 407 In the aftermath Siam had a seat at the Paris Peace Conference, and gained freedom of taxation and the revocation of extraterritoriality.[82]: 408
Constitutional monarchy, World War II and Cold War (1932–1975)

A bloodless revolution took place in 1932, in which Prajadhipok was forced to grant the country's first constitution, thereby ending centuries of feudal and absolute monarchy. The combined results of economic hardships brought on by the Great Depression, sharply falling rice prices, and a significant reduction in public spending caused discontent among aristocrats.[26]: 25 In 1933, a counter-revolutionary rebellion occurred which aimed to reinstate absolute monarchy, but failed.[82]: 446–8 Prajadhipok's conflict with the government eventually led to abdication. The government selected Ananda Mahidol, who was studying in Switzerland, to be the new king.[82]: 448–9
Later that decade, the army wing of Khana Ratsadon came to dominate Siamese politics. Plaek Phibunsongkhram who became premier in 1938, started political oppression and took an openly anti-royalist stance.[82]: 457 His government adopted nationalism and Westernisation, anti-Chinese and anti-French policies.[26]: 28
In 1939, there was a decree changing the name of the country from "Siam" to "Thailand". In 1941, Thailand was in a brief conflict with Vichy France, resulting in Thailand gaining some Lao and Cambodian territories.[82]: 462
On 8 December 1941, the Empire of Japan launched an invasion of Thailand, and fighting broke out shortly before Phibun ordered an armistice. Japan was granted free passage, and on 21 December Thailand and Japan signed a military alliance with a secret protocol, wherein the Japanese government agreed to help Thailand regain lost territories.[92] The Thai government then declared war on the United States and the United Kingdom.[82]: 465 The United Kingdom, whose colony Malaya was under immediate threat from Thai forces, responded in kind, but the United States refused to declare war and ignored Thailand's declaration.[93]: 66 The Free Thai Movement was launched both in Thailand and abroad to oppose the government and Japanese occupation.[82]: 465–6 After the war ended in 1945, Thailand signed formal agreements to end the state of war with the Allies.

In June 1946, young King Ananda was found dead under mysterious circumstances. His younger brother Bhumibol Adulyadej ascended to the throne. Thailand joined the Southeast Asia Treaty Organization (SEATO) to become an active ally of the United States in 1954.[82]: 493 Field Marshal Sarit Thanarat launched a coup in 1957, which removed Khana Ratsadon from politics. His rule (premiership 1959–1963) was autocratic; he built his legitimacy around the god-like status of the monarch and by channelling the government's loyalty to the king.[82]: 511 His government improved the country's infrastructure and education.[82]: 514 After the United States joined the Vietnam War in 1961, there was a secret agreement wherein the U.S. promised to protect Thailand.[82]: 523
The period brought about increasing modernisation and Westernisation of Thai society. Rapid urbanisation occurred when the rural populace sought work in growing cities. Rural farmers gained class consciousness and were sympathetic to the Communist Party of Thailand.[82]: 528 Economic development and education enabled the rise of a middle class in Bangkok and other cities.[82]: 534 In October 1971, there was a large demonstration against the dictatorship of Thanom Kittikachorn (premiership 1963–1973), which led to civilian casualties.[82]: 541–3 Bhumibol installed Sanya Dharmasakti (premiership 1973–1975) to replace him, marking the first time that the king had intervened in Thai politics directly since 1932.[94] The aftermath of the event marked a short-lived parliamentary democracy,[94] often called the "era when democracy blossomed" (ยุคประชาธิปไตยเบ่งบาน).[citation needed]
Contemporary history
Constant unrest and instability, as well as fear of a communist takeover after the fall of Saigon, made some ultra-right groups brand leftist students as communists.[82]: 548 This culminated in the Thammasat University massacre in October 1976.[82]: 548–9 A coup d'état on that day brought Thailand a new ultra-right government, which cracked down on media outlets, officials, and intellectuals, and fuelled the communist insurgency. Another coup the following year installed a more moderate government, which offered amnesty to communist fighters in 1978.[95]
Fuelled by Indochina refugee crisis, Vietnamese border raids and economic hardships, Prem Tinsulanonda became the Prime Minister from 1980 to 1988. The communists abandoned the insurgency by 1983. Prem's premiership was dubbed "semi-democracy" because the Parliament was composed of all elected House and all appointed Senate. The 1980s also saw increasing intervention in politics by the monarch, who rendered two coups in 1981 and 1985 attempts against Prem failed. In 1988 Thailand had its first elected prime minister since 1976.[96]
Suchinda Kraprayoon, who was the coup leader in 1991 and said he would not seek to become prime minister,[97] was nominated as one by the majority coalition government after the 1992 general election. This caused a popular demonstration in Bangkok, which ended with a bloody military crackdown. Bhumibol intervened in the event and signed an amnesty law, Suchinda then resigned.[98]
The 1997 Asian financial crisis originated in Thailand and ended the country's 40 years of uninterrupted economic growth.[99]: 3 Chuan Leekpai's government took an IMF loan with unpopular provisions.[100]
The 2004 Indian Ocean earthquake and tsunami hit the country, mostly in the south, claiming around 5,400 lives in Phuket, Phang Nga, Ranong, Krabi, Trang, and Satun, with thousands still missing.[101]
The populist Thai Rak Thai party, led by prime minister Thaksin Shinawatra, governed from 2001 until 2006. His policies were successful in reducing rural poverty[102] and initiated universal healthcare in the country.[103] However, Thaksin was viewed as a corrupt populist who was destroying the middle class in order to favor himself and the rural poor. He also faced criticism over his response to a South Thailand insurgency which escalated starting from 2004. Additionally, his recommendations to the rural poor directly conflicted with King Bhumibol's recommendations, drawing the ire of royalists, a powerful faction in Thailand. In response, the royalists made up a story about how Thaskin and his "advisors gathered in Finland to plot the overthrow of the monarchy". Meanwhile, massive protests against Thaksin led by the People's Alliance for Democracy (PAD) started in his second term as prime minister. Eventually, the monarchy and the military agree to oust the leader. In this case, the military first sought permission from the king to oust Thaksin, the permission was denied. But then, the king rejected Thaksin's choice to lead the army, allowing a military leader to be put into power who wanted the coup.1 Then, the army dissolved Thaksin's party with a coup d'état in 2006 and banned over a hundred of its executives from politics. After the coup, a military government was installed which lasted a year.[104][105]

Coming back to democracy was a process that took very active participation of the people. The people frequently stormed government buildings and the military threatened yet another coup.[104] Finally, in 2007, a civilian government led by the Thaksin-allied People's Power Party (PPP) was elected. Another protest led by PAD ended with the dissolution of PPP, and the Democrat Party led a coalition government in its place. The pro-Thaksin United Front for Democracy Against Dictatorship (UDD) protested both in 2009 and in 2010, the latter of which ended with a violent military crackdown causing more than 70 civilian deaths.[106]
After the general election of 2011, the populist Pheu Thai Party won a majority and Yingluck Shinawatra, Thaksin's younger sister, became prime minister. The People's Democratic Reform Committee organised another anti-Shinawatra protest[107] after the ruling party proposed an amnesty bill which would benefit Thaksin.[108] Yingluck dissolved parliament and a general election was scheduled, but was invalidated by the Constitutional Court. The crisis ended with another coup d'état in 2014.[109]
The ensuing National Council for Peace and Order, a military junta led by General Prayut Chan-o-cha, led the country until 2019. Civil and political rights were restricted, and the country saw a surge in lèse-majesté cases. Political opponents and dissenters were sent to "attitude adjustment" camps;[110] this was described by academics as showing the rise of fascism.[111] Bhumibol, the longest-reigning Thai king, died in 2016, and his son Vajiralongkorn ascended to the throne. The referendum and adoption of Thailand's current constitution happened under the junta's rule.[vi] The junta also bound future governments to a 20-year national strategy 'road map' it laid down, effectively locking the country into military-guided democracy.[113] In 2019, the junta agreed to schedule a general election in March.[110] Prayut continued his premiership with the support of Palang Pracharath Party-coalition in the House and junta-appointed Senate, amid allegations of election fraud.[114] The 2020–21 pro-democracy protests were triggered by increasing royal prerogative, democratic and economic regression from the Royal Thai Armed Forces supported by the monarchy in the wake of the coup d'état in 2014, dissolution of the pro-democracy Future Forward Party, distrust in the 2019 general election and the current political system, forced disappearance and deaths of political activists including Wanchalearm Satsaksit, and political corruption scandals,[115][116] which brought forward unprecedented demands to reform the monarchy[117] and the highest sense of republicanism in the country.[118]
In May 2023, Thailand's reformist opposition, the progressive Move Forward Party (MFP) and the populist Pheu Thai Party, won the general election, meaning the royalist-military parties that supported Prime Minister Prayuth Chan-ocha lost power.[119] On 22 August 2023, Srettha Thavisin of the populist Pheu Thai party, became Thailand's new prime minister, while the Pheu Thai party's billionaire figurehead Thaksin Shinawatra returned to Thailand after years in self-imposed exile.[120] Thavisin was later dismissed from his prime ministerial role on 14 August 2024 by the Constitutional Court for his "gross ethics violations."[121]
Geography

Totalling 513,120 square kilometres (198,120 sq mi), Thailand is the 50th-largest country by total area.[1] Thailand comprises several distinct geographic regions, partly corresponding to the provincial groups. The north of the country is the mountainous area of the Thai highlands, with the highest point being Doi Inthanon in the Thanon Thong Chai Range at 2,565 metres (8,415 ft) above sea level. The northeast, Isan, consists of the Khorat Plateau, bordered to the east by the Mekong River. The centre of the country is dominated by the predominantly flat Chao Phraya river valley, which runs into the Gulf of Thailand. Southern Thailand consists of the narrow Kra Isthmus that widens into the Malay Peninsula.
The Chao Phraya and the Mekong River are the indispensable water courses of rural Thailand. Industrial scale production of crops use both rivers and their tributaries. The Gulf of Thailand covers 320,000 square kilometres (124,000 sq mi) and is fed by the Chao Phraya, Mae Klong, Bang Pakong, and Tapi Rivers. It contributes to the tourism sector owing to its clear shallow waters along the coasts in the southern region and the Kra Isthmus. The eastern shore of the Gulf of Thailand has the kingdom's premier deepwater port in Sattahip and its busiest commercial port, Laem Chabang. Phuket, Krabi, Ranong, Phang Nga and Trang, and their islands, all lay along the coasts of the Andaman Sea.[citation needed]
Climate

Thailand's climate is influenced by monsoon winds that have a seasonal character (the southwest and northeast monsoon).[122]: 2 Most of the country is classified as Köppen's tropical savanna climate.[123] The majority of the south as well as the eastern tip of the east have a tropical monsoon climate. Parts of the south also have a tropical rainforest climate.
A year in Thailand is divided into three seasons.[122]: 2 The first is the rainy or southwest monsoon season (mid–May to mid–October), which is caused by southwestern wind from the Indian Ocean.[122]: 2 Rainfall is also contributed by Intertropical Convergence Zone (ITCZ) and tropical cyclones,[122]: 2 with August and September being the wettest period of the year.[122]: 2 The country receives a mean annual rainfall of 1,200 to 1,600 mm (47 to 63 in).[122]: 4 Winter or the northeast monsoon occurs from mid–October until mid–February.[122]: 2 Most of Thailand experiences dry weather with mild temperatures.[122]: 2, 4 Summer or the pre–monsoon season runs from mid–February until mid–May.[122]: 3
Due to their inland position and latitude, the north, northeast, central and eastern parts of Thailand experience a long period of warm weather, where temperatures can reach up to 40 °C (104 °F) during March to May,[122]: 3 in contrast to close to or below 0 °C (32 °F) in some areas in winter.[122]: 3 Southern Thailand is characterised by mild weather year-round with less diurnal and seasonal variations in temperatures due to maritime influences.[122]: 3 It receives abundant rainfall, particularly during October to November.[122]: 2 Thailand is among the world's ten countries that are most exposed to climate change. In particular, it is highly vulnerable to rising sea levels and extreme weather events.[124][125]
Biodiversity and conservation

National parks in Thailand are defined as an area that contains natural resources of ecological importance or unique beauty, or flora and fauna of special importance. Thailand's protected areas include 156 national parks, 58 wildlife sanctuaries, 67 non-hunting areas, and 120 forest parks. They cover almost 31 per cent of the kingdom's territory.[127] The parks are administered by the National Parks, Wildlife and Plant Conservation Department (DNP) of the Ministry of Natural Resources and Environment (MNRE).
Thailand has a mediocre but improving performance in the global Environmental Performance Index (EPI) with an overall ranking of 91 out of 180 countries in 2016. The environmental areas where Thailand performs worst (i.e., highest-ranking) are air quality (167), environmental effects of the agricultural industry (106), and the climate and energy sector (93), the later mainly because of a high CO2 emission per kWh produced. Thailand performs best (i.e., lowest-ranking) in water resource management (66), with some major improvements expected for the future, and sanitation (68).[128][129] The country had a 2019 Forest Landscape Integrity Index mean score of 6.00/10, ranking it 88th globally out of 172 countries.[130]
The population of elephants, the country's national symbol, has fallen from 100,000 in 1850 to an estimated 2,000.[126] Poachers have long hunted elephants for ivory and hides, and now increasingly for meat.[131] Young elephants are often captured for use in tourist attractions or as work animals, where there have been claims of mistreatment.[132] In 1989, the government banned the use of elephants for logging, leading many elephant owners to move their domesticated animals to the tourism industry.[133]
Poaching of protected species remains a major problem. Tigers, leopards, and other large cats are hunted for their pelts. Many are farmed or hunted for their meat, which supposedly has medicinal properties. Although such trade is illegal, the well-known Bangkok market Chatuchak is still known for the sale of endangered species.[134] The practice of keeping wild animals as pets affects species such as Asiatic black bear, Malayan sun bear, white-handed lar, pileated gibbon, and binturong.[135]
Politics and government
_(cropped).png)

Prior to 1932, Thai kings were feudal or absolute monarchs. During Sukhothai Kingdom, the king was seen as a Dharmaraja or 'king who rules in accordance with Dharma'. The system of government was a network of tributaries ruled by local lords. Modern absolute monarchy and statehood was established by Chulalongkorn when he transformed the decentralized protectorate system into a unitary state. On 24 June 1932, Khana Ratsadon (People's Party) carried out a bloodless revolution which marked the beginning of constitutional monarchy.
Thailand has had 20 constitutions and charters since 1932, including the latest and current 2017 Constitution. All constitutions state that the politics is conducted within the framework of a constitutional monarchy, but the de facto form of government has ranged from military dictatorship to electoral democracy.[136][137] Thailand's current form of government is part democracy and part dictatorship; many terms are used to describe it.[vii] Thailand has had the fourth-most coups in the world.[143] "Uniformed or ex-military men have led Thailand for 55 of the 83 years" between 1932 and 2009.[144] Most recently, the military junta self-titled as the National Council for Peace and Order ruled the country between 2014 and 2019.

Government is separated into three branches:
- The legislative branch: the National Assembly is composed of the Senate, the 200-member indirectly elected upper house and House of Representatives, the elected 500-member lower house. Its most recent election is the 2023 general election. The coalition led by Pheu Thai Party currently holds the majority. The 2024 Thai Senate election was the first senate election held under the current constitution in the process criticized as "the most complicated election in the world."[145] The senate is allegedly dominated by Bhumjaithai Party-affiliated senators.[146]
- The executive branch consisting of the Prime Minister of Thailand, the head of government, and other cabinet members of up to 35 people. The Prime Minister was elected by the National Assembly. The current constitution mandates that prime ministers are to be considered from candidates nominated by political parties before the election. The current prime minister is Paetongtarn Shinawatra, a member of the Pheu Thai Party.
- The judiciary is supposed to be independent of the executive and the legislative branches, although judicial rulings are suspected of being based on political considerations rather than on existing law.[147]
Military and bureaucratic aristocrats fully controlled political parties between 1946 and the 1980s.[148]: 16 Most parties in Thailand are short-lived.[149]: 246 Between 1992 and 2006, Thailand had a two-party system.[149]: 245 Later constitutions created a multi-party system where a single party cannot gain a majority in the house.
A hereditary monarch serves as Thailand's head of state. The current King of Thailand is Vajiralongkorn (Rama X), who has reigned since October 2016. The powers of the king are limited by the constitution and he is primarily a symbolic figurehead. However, the monarch still occasionally intervenes in Thai politics, as all constitutions pave the way for customary royal rulings. Some academics outside Thailand, including Duncan McCargo and Federico Ferrara, noted extraconstitutional role of the monarch through a "network monarchy" behind the political scenes.[150] The monarchy is protected by the severe lèse majesté law, even though the people's attitude towards the institution varies from one reign to another.[151][152]
The kings are protected by lèse-majesté laws which allow critics to be jailed for three to fifteen years.[153] After the coup d'état in 2014, Thailand had the highest number of lèse-majesté prisoners in the nation's history.[154][155] Human rights in Thailand has been rated not free on the Freedom House Index since 2014.[156] On August 7, 2024, Thailand's Constitutional Court banned the victors of the 2023 parliamentary elections, the Move Forward Party and all of its leaders from politics for its proposal to reform the lèse-majesté law, arguing it posed a threat to the constitutional order.[157] The Economist criticized the move as an example of "lawfare" and pointed to the dissolution of its predecessor party, Future Forward in 2020, as the latest example of how an "alliance of conservative forces in Thailand—including monarchists, the army and a handful of business tycoons—has sought to suppress opposition".[157][158]
On the Freedom in the World 2024 Report for Thailand, their status improved from not free to partly free due to competitive parliamentary elections and the formation of a new governing coalition by what had been a major opposition party, though unelected senators ensured that the party with the most votes was excluded.[159]
Administrative divisions
Thailand is a unitary state; the administrative services of the executive branch are divided into three levels by National Government Organisation Act, BE 2534 (1991): central, provincial and local. Thailand is composed of 76 provinces (จังหวัด, changwat),[160] which are first-level administrative divisions. There are also two specially governed districts: the capital Bangkok and Pattaya. Bangkok is at provincial level and thus often counted as a province. Each province is divided into districts (อำเภอ, amphoe) and the districts are further divided into sub-districts (ตำบล, tambons). The name of each province's capital city (เมือง, mueang) is the same as that of the province. For example, the capital of Chiang Mai Province (Changwat Chiang Mai) is Mueang Chiang Mai or Chiang Mai. All provincial governors and district chiefs, which are administrators of provinces and districts respectively, are appointed by the central government.[161] Thailand's provinces are sometimes grouped into four to six regions, depending on the source.
Foreign relations

Siam's and Thailand's way of conducting foreign relations has long been described as "bamboo bending with the wind", of policies that are "always solidly rooted, but flexible enough to bend whichever way the wind blows in order to survive,"[162] or adaptable and pragmatic. In order to secure independence, it sought to pit one great power against the others so that it would be dominated by none.[163]
During the Cold War, Thailand sought to prevent the spread of communism so it joined the United States, including participating in SEATO alliance, sending expeditions to Korea and Vietnam, and offering the US to use its base. Thailand is one of the five founding members of Association of Southeast Asian Nations (ASEAN), initially to safeguard against communism. The end of Vietnam War was a turning point in Thai foreign policy and afterwards it sought to improve relations with Communist China and its now-Communist neighbours. Thailand remains an active member of ASEAN and seek to project its influence in it. Thailand has developed increasingly close ties with other members, with progressing regional co-operation in economic, trade, banking, political, and cultural matters.[164]
In the 2000s, Thailand had taken an active role on the international stage and participated fully in international and regional organisations. It is a major non-NATO ally and Priority Watch List Special 301 Report of the United States. When East Timor gained independence from Indonesia, Thailand contributed troops to the international peacekeeping effort.[165] As part of its effort to increase international ties, Thailand had reached out to such regional organisations as the Organization of American States (OAS)[166] and the Organization for Security and Co-operation in Europe (OSCE).[167]
During Thaksin Shinawatra's premiership, negotiations for several free trade agreements with China, Australia, Bahrain, India, and the US were initiated. Thaksin sought to position Thailand as a regional leader, initiating various development projects in poorer neighbouring countries. More controversially, he established close, friendly ties with the Burmese dictatorship.[168] Thailand joined the US-led invasion of Iraq, sending a humanitarian contingent until September 2004.[169] Thailand also had contributed troops to reconstruction efforts in Afghanistan.[170]
In April 2009, the Cambodian–Thai border dispute brought troops on territory immediately adjacent to the 900-year-old ruins of Cambodia's Preah Vihear Hindu temple near the border.[171][172]
After the 2014 coup, Thailand leaned more towards China.[173] Growing Chinese influence and capital inflow caused some members of parliament to raise the concern about "economic colony" under China after many concessions.[174]
During the Israel-Hamas military conflict in 2023, at first Thailand's prime minister stated that his government strongly condemns the attack against Israel and extends its deepest condolences to the government and the people of Israel[175] but the government later changed its position and announced that Thailand adopted a neutral stance in this conflict.[176] 28 Thai nationals were killed in this conflict.[177]
Armed forces

The Royal Thai Armed Forces (กองทัพไทย; RTGS: Kong Thap Thai) constitute the military of the Kingdom of Thailand. It consists of the Royal Thai Army (กองทัพบกไทย), the Royal Thai Navy (กองทัพเรือไทย), and the Royal Thai Air Force (กองทัพอากาศไทย). It also incorporates various paramilitary forces.[citation needed]
The Thai Armed Forces have a combined manpower of 306,000 active duty personnel and another 245,000 active reserve personnel.[178] The head of the Thai Armed Forces (จอมทัพไทย, Chom Thap Thai) is the king,[179] although this position is only nominal. The armed forces are managed by the Ministry of Defence of Thailand, which is headed by the Minister of Defence (a member of the cabinet of Thailand) and commanded by the Royal Thai Armed Forces Headquarters, which in turn is headed by the Chief of Defence Forces of Thailand.[180] Thai annual defense budget almost tripled from US$1.98 billion in 2005 to US$5.88 billion in 2016, accounting for approximately 1.4% of GDP.[181] Thailand ranked 16th worldwide in the Military Strength Index based on the Credit Suisse report in September 2015.[182]
.jpg)
The military is also tasked with humanitarian missions, such as escorting Rohingya to Malaysia or Indonesia,[183] ensuring security and welfare for refugees during Indochina refugee crisis.[184]
According to the constitution, serving in the armed forces is a duty of all Thai citizens.[185] Thailand still use active draft system for males over the age of 21. They are subjected to varying lengths of active service depending on the duration of reserve training as Territorial Defence Student and their level of education. Those who have completed three years or more of reserve training will be exempted entirely. The practice has long been criticized, as some media question its efficacy and value.[186][187] It is alleged that conscripts end up as servants to senior officers[188] or clerks in military cooperative shops.[189][190] In a report issued in March 2020, Amnesty International charged that Thai military conscripts face institutionalised abuse systematically hushed up by military authorities.[191]
Critics observed that Thai military's main objective is to deal with internal rather than external threats.[192] Internal Security Operations Command is called the political arm of the Thai military, which has overlapping social and political functions with civilian bureaucracy. It also has anti-democracy mission.[192] The military is also notorious for numerous corruption incidents, such as accusation of human trafficking,[193] and nepotism in promotion of high-ranking officers.[194] The military is deeply entrenched in politics. Most recently, the appointed senators include more than 100 active and retired military.[195]
Thailand is the 75th most peaceful country in the world, according to the 2024 Global Peace Index.[196]
Economy
| Nominal GDP | ฿14.53 trillion (2016)[197] |
|---|---|
| GDP growth | 3.9% (2017)[198] |
| Headline inflation | 0.7% (2017)[198] |
| Core inflation | 0.6% (2017)[198] |
| Employment-to-population ratio | 68.0% (2017)[199]: 29 |
| Unemployment | 1.2% (2017)[198] |
| Total public debt | ฿6.37 trillion (Dec. 2017)[200] |
| Poverty | 8.61% (2016)[199]: 36 |
| Net household worth | ฿20.34 trillion (2010)[201]: 2 |
.jpeg)
The economy of Thailand is heavily export-dependent, with exports accounting for more than two-thirds of gross domestic product (GDP). Thailand exports over US$105 billion worth of goods and services annually.[1] Major exports include cars, computers, electrical appliances, rice, textiles and footwear, fishery products, rubber, and jewellery.[1]
Thailand is an emerging economy and is considered a newly industrialised country. Thailand had a 2017 GDP of US$1.236 trillion (on a purchasing power parity basis).[202] Thailand is the second largest economy in Southeast Asia after Indonesia. Thailand ranks midway in the wealth spread in Southeast Asia as it is the fourth richest nation according to GDP per capita, after Singapore, Brunei, and Malaysia.
Thailand functions as an anchor economy for the neighbouring developing economies of Laos, Myanmar, and Cambodia. In the third quarter of 2014, the unemployment rate in Thailand stood at 0.84% according to Thailand's National Economic and Social Development Board (NESDB).[203]
In 2017, the Thai economy grew an inflation-adjusted 3.9%, up from 3.3% in 2016, marking its fastest expansion since 2012.[204] High public spending, especially during the COVID-19 pandemic, prompted the authorities to raise Thailand's public debt ceiling from 60% to 70% of GDP.[205] As of 2024[update], Thailand struggle with low productivity, poor education, high household debt, low private investment and slow economic growth,[206][207] with an economic research group forecasted an annual GDP growth of below 2% in the next decades without structural reforms.[208]
Income and wealth disparities

Thais have median wealth per one adult person of $1,469 in 2016,[209]: 98 increasing from $605 in 2010.[209]: 34 In 2016, Thailand was ranked 87th in Human Development Index, and 70th in the inequality-adjusted HDI.[210]
In 2017, Thailand's median household income was ฿26,946 per month.[211]: 1 Top quintile households had a 45.0% share of all income, while bottom quintile households had 7.1%.[211]: 4 There were 26.9 million persons who had the bottom 40% of income earning less than ฿5,344 per person per month.[212]: 5 During the 2013–2014 Thai political crisis, a survey found that anti-government PDRC mostly (32%) had a monthly income of more than ฿50,000, while pro-government UDD mostly (27%) had between ฿10,000 and ฿20,000.[213]: 7
In 2014, Credit Suisse reported that Thailand was the world's third most unequal country, behind Russia and India.[214] The top 10% richest held 79% of the country's assets.[214] The top 1% held 58% of the assets.[214] The 50 richest Thai families had a total net worth accounting to 30% of GDP.[214] Bank of Thailand reported that during 2006–16, Thailand's top 5% largest companies had 85% of all corporate revenue in the nation, and only 6% of the country's companies were in export industries, which made up 60% of the country's GDP.[215]
In 2016, 5.81 million people lived in poverty, or 11.6 million people (17.2% of population) if "near poor" is included.[212]: 1 The proportion of the poor relative to total population in each region was 12.96% in the Northeast, 12.35% in the South, and 9.83% in the North.[212]: 2 In 2017, there were 14 million people who applied for social welfare (yearly income of less than ฿100,000 was required).[214] In the first quarter of 2023, Thai household debts totaled 14.6 trillion baht or 89.2% of GDP; the average debt per household was approximately 500,000 baht.[216] In 2016, there were estimated 30,000 homeless persons in the country.[217]
Exports and manufacturing
The economy of Thailand is heavily export-dependent, with exports accounting for more than two-thirds of gross domestic products (GDPs). Major exports include cars, computers, electrical appliances, rice, textiles and footwear, fishery products, rubber, and jewellery.[1] In 2022, Thailand's export of goods is worth roughly US$290 billion while its import worth roughly US$305 billion.[218]
Substantial industries include electric appliances, components, computer components, and vehicles. Thailand's recovery from the 1997–1998 Asian financial crisis depended mainly on exports, among various other factors. As of 2012[update], the Thai automotive industry was the largest in Southeast Asia and the 9th largest in the world.[219][220][221] The Thailand industry has an annual output of near 1.5 million vehicles, mostly commercial vehicles.[221]
Most of the vehicles built in Thailand are developed and licensed by foreign producers, mainly Japanese and American. The Thai car industry takes advantage of the ASEAN Free Trade Area (AFTA) to find a market for many of its products. Eight manufacturers, five Japanese, two US, and Tata of India, produce pick-up trucks in Thailand.[222] As of 2012, due to its favorable taxation for 2-door pick-ups at only 3–12% against 17–50% for passenger cars, Thailand was the second largest consumer of pick-up trucks in the world, after the US.[223] In 2014, pick-ups accounted for 42% of all new vehicle sales in Thailand.[222]
Tourism

Tourism makes up about 6% of the country's economy. Prior to the pandemic, Thailand was the world's eighth most visited country according to the World Tourism rankings compiled by the United Nations World Tourism Organization. In 2019, Thailand received 39.8 million international tourists, ahead of United Kingdom and Germany[224] and was the fourth highest in international tourism earning 60.5 billion US dollars.
Thailand was the most visited country in Southeast Asia in 2013, according to the World Tourism Organization. Estimates of tourism receipts directly contributing to the Thai GDP of 12 trillion baht range from 9 per cent (1 trillion baht) (2013) to 16 per cent.[225] When including the indirect effects of tourism, it is said to account for 20.2 per cent (2.4 trillion baht) of Thailand's GDP.[226]: 1
Asian tourists primarily visit Thailand for Bangkok and the historical, natural, and cultural sights in its vicinity. Western tourists not only visit Bangkok and surrounding areas; many travel to the southern beaches and islands. The north is the chief destination for trekking and adventure travel with its diverse ethnic minority groups and forested mountains. The region hosting the fewest tourists is Isan. To accommodate foreign visitors, a separate tourism police with offices were set up in the major tourist areas and an emergency telephone number.[227]
Thailand ranks as the worlds fifth largest medical tourism destination in spending, according to the World Travel and Tourism Council, attracting over 2.5 million visitors in 2018,[228] and is number one in Asia.[229] The country is popular for the growing practice of sex reassignment surgery (SRS) and cosmetic surgery. In 2010–2012, more than 90% of medical tourists traveled to Thailand for SRS.[230] Prostitution in Thailand and sex tourism also form a de facto part of the economy. Campaigns promote Thailand as exotic to attract tourists.[231] One estimate published in 2003 placed the trade at US$4.3 billion per year or about 3% of the Thai economy.[232] It is believed that at least 10% of tourist dollars are spent on the sex trade.[233]
Agriculture and natural resources

Forty-nine per cent of Thailand's labour force is employed in agriculture.[234] This is down from 70% in 1980.[234] Rice is the most important crop in the country and Thailand had long been the world's leading exporter of rice, until recently falling behind both India and Vietnam.[235] Thailand has the highest percentage of arable land, 27.25%, of any state in the Greater Mekong Subregion.[236] About 55% of the arable land area is used for rice production.[237]
Agriculture has been experiencing a transition from labour-intensive and transitional methods to a more industrialised and competitive sector.[234] Between 1962 and 1983, the agricultural sector grew by 4.1% per year on average and continued to grow at 2.2% between 1983 and 2007.[234] The relative contribution of agriculture to GDP has declined while exports of goods and services have increased.
Furthermore, access to biocapacity in Thailand is lower than world average. In 2016, Thailand had 1.2 global hectares[238] of biocapacity per person within its territory, a little less than world average of 1.6 global hectares per person.[239] In contrast, in 2016, they used 2.5 global hectares of biocapacity—their ecological footprint of consumption. This means they use about twice as much biocapacity as Thailand contains, resulting in a deficit.[238]
Informal economy
.jpg)
In 2012, it was estimated that informal workers comprised 62.6% of the Thai workforce. The Ministry of Labour defines informal workers to be individuals who work in informal economies and do not have employee status under a given country's Labour Protection Act (LPA). The informal sector in Thailand has grown significantly over the past 60 years over the course of Thailand's gradual transition from an agriculture-based economy to becoming more industrialised and service-oriented.[240] Between 1993 and 1995, ten per cent of the Thai labour force moved from the agricultural sector to urban and industrial jobs, especially in the manufacturing sector. It is estimated that between 1988 and 1995, the number of factory workers in the country doubled from two to four million, as Thailand's GDP tripled.[241]
While the Asian financial crisis that followed in 1997 hit the Thai economy hard, the industrial sector continued to expand under widespread deregulation, as Thailand was mandated to adopt a range of structural adjustment reforms upon receiving funding from the IMF and World Bank. These reforms implemented an agenda of increased privatisation and trade liberalisation in the country, and decreased federal subsidisation of public goods and utilities, agricultural price supports, and regulations on fair wages and labour conditions.[242][full citation needed] Many migrant farmers took jobs in sweatshops and factories with few labour regulations and often exploitative conditions.[243] Those that could not find formal factory work, including illegal migrants and the families of rural Thai migrants, are under the regulation imposed by the structural adjustment programs. Scholars argue that the economic consequences and social costs of Thailand's labour reforms in the wake of the 1997 Asian financial crisis fell on individuals and families rather than the state.[242]

Informal labour in entertainment, nightlife, and the sex industry face additional vulnerabilities, including recruitment into circles of sexual exploitation and human trafficking.[240] A 2012 study found that 64% of informal workers had not completed education beyond primary school. Many informal workers are also migrants, only some of which have legal status in the country. The informal labour sector is also not recognised under the Labour Protection Act (LPA). Thai social security policies fail to protect against workplace accidents and unemployment and retirement insurance. Many informal workers are not legally contracted for their employment, and many do not make a living wage.[240] Tens of thousands of migrants from neighboring countries face exploitation in a few industries,[244] especially in fishing where slave-like conditions have been reported.[245]
Science and technology
Thailand ranked 41st in the Global Innovation Index in 2024.[246] The Ministry of Higher Education, Science, Research and Innovation and its agencies oversees the development of science, technology, and research in Thailand. According to the National Research Council of Thailand, the country devoted 1.1% of its GDP to the research and development of science in 2019, with over 166,788 research and development personnel in full-time equivalent that year.[247][248]
Infrastructure
Transportation

The State Railway of Thailand (SRT) operates all of Thailand's national rail lines. Krung Thep Aphiwat Central Terminal and Bangkok (Hua Lamphong) are the main termini of intercity routes. Phahonyothin and ICD Lat Krabang are the main freight terminals. As of 2024[update] SRT had 4,507 km (2,801 mi) of track, all of it meter gauge. Nearly all is single-track (2,847.1 km), although some important sections around Bangkok are double (1,089.9 km or 677.2 mi) or triple-tracked (107 km or 66 mi), and there are plans to extend this.[249][250]
Rail transport in Bangkok includes long-distance services. There are four rapid transit rail systems in the capital: the BTS Skytrain, MRT, SRT Red Lines, and the Airport Rail Link.[251] In Bangkok, there were two failed rapid rail projects Lavalin Skytrain and Bangkok Elevated Road and Train System, before Mass Rapid Transit Master Plan in Bangkok Metropolitan Region was endorsed by the cabinet on 27 September 1994 and implemented from 1995 to the present.[252]
Thailand has 390,000 kilometres (240,000 miles) of highways.[253] As of 2017[update], Thailand has over 462,133 roads and 37 million registered vehicles, 20 million of them motorbikes.[254] A number of undivided two-lane highways have been converted into divided four-lane highways. Within the Bangkok Metropolitan Region, there are a number of controlled-access highways. There are 4,125 public vans operating on 114 routes from Bangkok alone.[255] Other forms of road transport includes tuk-tuks, taxis—with over 80,647 registered taxis nationwide as of 2018,[256] vans (minibus), motorbike taxis, and songthaews.
As of 2012[update], Thailand has 103 airports with 63 paved runways, in addition to 6 heliports. The busiest airport in the country is Bangkok's Suvarnabhumi Airport.[257]
Energy
75% of Thailand's electrical generation is powered by natural gas in 2014.[258] Coal-fired power plants produce an additional 20% of electricity, with the remainder coming from biomass, hydro, and biogas.[258] Compared to other ASEAN's countries, Thailand is the largest importer of gas in weight.[259]: 17 In 2022, Thailand's oil and gas production dropped by 19% and 17%, respectively.[260][259]: 8
The government, in 2018, has developed an Alternative Energy Development Plan 2018–2037 (AEDP 2018). The plan defines goals for the increase of renewable energy to almost 30,000 MW by 2037.[261][262]
Demographics
Ethnic groups
of Thailand
(2015 estimate by The World Factbook)[1]
Thailand has an estimated population of 71.7 million as of 2023;[263][contradictory] Thailand's first census in 1909 found the population to be 8.2 million.[264] Thailand's population is largely rural, concentrated in the rice-growing areas of the central, northeastern, and northern regions. About 44.2% of Thailand's population lived in urban areas as of 2010[update], slowly increasing from 29.4% in the 1990 census and 31.1% in the 2000 census.[265]
Thailand's government-sponsored family planning program resulted in a dramatic decline in population growth from 3.1% in 1960 to around 0.4% today. In 1970, an average of 5.7 people lived in a Thai household; in 2022, the average Thai household size was 3 people.[266] Now, more than 20% of its population is aged over 60 and has a low birth rate, posing economic challenges.[267] The sex ratio between male and female is 1.05, with Thailand having slightly more males.[268]
Ethnic groups
.jpg)
As of 2010, Thai people make up the majority of Thailand's population (95.9%). The remaining 4.1% of the population are Burmese (2.0%), others (1.3%), and unspecified (0.9%).[1]
According to genetic research, the present-day Thai people were divided into three groups: the northern group (Khon Mueang) are closely related to the Tai ethnic groups in southern China, the northeastern group (Isan people) are mixed Tai and several Austroasiatic-speaking ethnic groups, while the central and southern groups (formerly called Siamese) strongly share genetic profiles with the Mon people.[37][38][39]
According to the Royal Thai Government's 2011 Country Report to the UN Committee responsible for the International Convention for the Elimination of All Forms of Racial Discrimination, available from the Department of Rights and Liberties Promotion of the Thai Ministry of Justice,: 3 62 ethnic communities are officially recognized in Thailand. Twenty million Central Thai (together with approximately 650,000 Khorat Thai) made up approximately 20,650,000 (34.1 per cent) of the state's population of 60,544,937[269] at the time of completion of the Mahidol University Ethnolinguistic Maps of Thailand data (1997).[270]
The 2011 Thailand Country Report provides population numbers for mountain peoples ('hill tribes') and ethnic communities in the Northeast and is explicit about its main reliance on the Mahidol University Ethnolinguistic Maps of Thailand data.[270] Thus, though over 3.288 million people in the Northeast alone could not be categorised, the population and percentages of other ethnic communities c. 1997 are known for all of Thailand and constitute minimum populations. In descending order, the largest (equal to or greater than 400,000) are a) 15,080,000 Lao (24.9 per cent) consisting of the Thai Lao (14 million) and other smaller Lao groups, namely the Thai Loei (400–500,000), Lao Lom (350,000), Lao Wiang/Klang (200,000), Lao Khrang (90,000), Lao Ngaew (30,000), and Lao Ti (10,000); b) six million Khon Muang (9.9 per cent, also called Northern Thais); c) 4.5 million Pak Tai (7.5 per cent, also called Southern Thais); d) 1.4 million Khmer Leu (2.3 per cent, also called Northern Khmer); e) 900,000 Malay (1.5%); f) 500,000 Nyaw (0.8 per cent); g) 470,000 Phu Thai (0.8 per cent); h) 400,000 Kuy/Kuay (also known as Suay) (0.7 per cent), and i) 350,000 Karen (0.6 per cent).: 7–13 Thai Chinese, those of significant Chinese heritage, are 14% of the population, while Thais with partial Chinese ancestry comprise up to 40% of the population.[271] Thai Malays represent 3% of the population, with the remainder consisting of Mons, Khmers, and various "hill tribes".[citation needed]
Increasing numbers of migrants from neighbouring Myanmar, Laos, and Cambodia, as well as from Nepal and India, have pushed the total number of non-national residents to around 3.5 million as of 2009[update], up from an estimated 2 million in 2008.[272] Some 41,000 Britons and 20,000 Australians live in Thailand.[273][274]
Population centres
| Rank | Name | Province | Pop. | Rank | Name | Province | Pop. | ||
|---|---|---|---|---|---|---|---|---|---|
_cropped.jpg) Bangkok  Nonthaburi City |
1 | Bangkok | Bangkok | 5,588,222 | 11 | Khon Kaen City | Khon Kaen | 110,615 |  Pak Kret City  Hat Yai City |
| 2 | Nonthaburi City | Nonthaburi | 251,026 | 12 | Nakhon Si Thammarat City | Nakhon Si Thammarat | 100,416 | ||
| 3 | Pak Kret City | Nonthaburi | 189,458 | 13 | Laem Chabang City | Chonburi | 89,457 | ||
| 4 | Hat Yai City | Songkhla | 149,459 | 14 | Rangsit City | Pathum Thani | 84,268 | ||
| 5 | Chaophraya Surasak City | Chonburi | 146,474 | 15 | Nakhon Sawan City | Nakhon Sawan | 81,239 | ||
| 6 | Surat Thani City | Surat Thani | 131,599 | 16 | Phuket City | Phuket | 77,778 | ||
| 7 | Nakhon Ratchasima City | Nakhon Ratchasima | 122,730 | 17 | Chiang Rai City | Chiang Rai | 77,545 | ||
| 8 | Chiang Mai City | Chiang Mai | 122,627 | 18 | Ubon Ratchathani City | Ubon Ratchathani | 72,855 | ||
| 9 | Udon Thani City | Udon Thani | 120,202 | 19 | Nakhon Pathom City | Nakhon Pathom | 72,753 | ||
| 10 | Pattaya City | Chonburi | 117,606 | 20 | Ko Samui City | Surat Thani | 68,994 | ||
Largest cities by urban population in Thailand
| |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Rank | Name | Province | Pop. | ||||||
.jpg) Bangkok Chiang Mai |
1 | Bangkok | Bangkok | 10,539,000 |  Khon Kaen | ||||
| 2 | Chiang Mai | Chiang Mai | 1,198,000 | ||||||
| 3 | Nakhon Ratchasima | Nakhon Ratchasima | 466,098 | ||||||
| 4 | Khon Kaen | Khon Kaen | 412,758 | ||||||
| 5 | Hat Yai | Songhkla | 404,044 | ||||||
| 6 | Udon Thani | Udon Thani | 400,581 | ||||||
| 7 | Chonburi | Chonburi | 342,959 | ||||||
| 8 | Pattaya | Chonburi | 328,961 | ||||||
| 9 | Si Racha | Chonburi | 327,172 | ||||||
| 10 | Phitsanulok | Phitsanulok | 281,929 | ||||||
Language

.jpg)
The official language of Thailand is Thai, a Kra–Dai language closely related to Lao, Shan in Myanmar, and numerous smaller languages spoken in an arc from Hainan and Yunnan south to the Chinese border. It is the principal language of education and government and spoken throughout the country.[275] The standard is based on the dialect of the central Thai people, and it is written in the Thai alphabet, an abugida script that evolved from the Khmer alphabet.[276] Sixty-two languages were recognised by the Royal Thai Government.[277] For the purposes of the national census, four dialects of Thai exist; these partly coincide with regional designations, such as Southern Thai and Northern Thai.[275]
The largest of Thailand's minority languages is the Lao dialect of Isan spoken in the northeastern provinces. In the far south, Kelantan-Pattani Malay is the primary language of Malay Muslims. Varieties of Chinese are also spoken by the large Thai Chinese population, with the Teochew dialect best-represented. Numerous tribal languages are also spoken, including many Austroasiatic languages such as Mon, Khmer, and Mlabri; Austronesian languages such as Cham, Moken and Urak Lawoi'; Sino-Tibetan languages like Lawa, Akha, and Karen; and other Tai languages such as Phu Thai, and Saek. Hmong is a member of the Hmong–Mien languages, which is now regarded as a language family of its own.[278][275]
Religion
The country's most prevalent religion is Theravada Buddhism, which is an integral part of Thai identity and culture. Active participation in Buddhism is among the highest in the world. Thailand has the second-largest number of Buddhists in the world after China.[279] According to the 2018 National Statistical Office data, 93.46% of the country's population self-identified as Buddhists.[2]

Muslims constitute the second largest religious group in Thailand, comprising 5.37% of the population in 2018.[2] Islam is concentrated mostly in the country's southernmost provinces: Pattani, Yala, Satun, Narathiwat, and part of Songkhla Chumphon, which are predominantly Malay, most of whom are Sunni Muslims. Christians represented 1.13% of the population in 2018, with the remaining population consisting of Hindus and Sikhs, who live mostly in the country's cities. There is also a small Jewish community in Thailand dating back to the 17th century.[280]
The constitution does not name an official state religion, and provides for freedom of religion. There have been no widespread reports of societal abuses or discrimination based on religious belief or practice.[281] Thai law officially recognizes five religious groups: Buddhists, Muslims, Brahmin-Hindus, Sikhs, and Christians.[282] However, some laws are inspired from Buddhist practices, such as banning alcohol sales on religious holidays.[283]
Education

In 1995, as minister of education, Sukavich Rangsitpollaid let out his plans for educational reform in Thailand.[284] The reform was considered a landmark movement after nearly 100 years of education under the previous system.[285] Thailand's youth literacy rate was 98.1% in 2015.[286] Education is provided by a school system of kindergartens, primary, lower secondary and upper secondary schools, numerous vocational colleges, and universities. Education is compulsory up to and including age 14, while the government is mandated to provide free education through to age 17. Issues concerning university entrance have been in constant upheaval for a number of years. The country is also one of the few that still mandates uniform up to the university years, which is still a subject of ongoing debate.[287]
In 2013, the Ministry of Information and Communication Technology announced that 27,231 schools would receive classroom-level access to high-speed internet.[288] However, the country's educational infrastructure was still underprepared for online teaching, as smaller and more remote schools were particularly hindered by COVID-19 restrictions.[289]
The number of higher education institutions in Thailand has grown over the past decades to 156 officially. The two top-ranking universities in Thailand are Chulalongkorn University and Mahidol University.[290] Thai universities' research output is still relatively low, even though the country's journal publications increased by 20% between 2011 and 2016.[291] Thailand has the second highest number of English-medium private international schools in Southeast Asian Nations.[292] Cram schools are especially popular for university entrance exams.[293]
Students in ethnic minority areas score consistently lower in standardised national and international tests.[294][295][296] This is likely due to unequal allocation of educational resources, weak teacher training, poverty, and low Thai language skill, the language of the tests.[294][297][298] As of 2020[update], Thailand was ranked 89th out of 100 countries globally for English proficiency.[299] Thailand is the third most popular study destination in ASEAN. The number of international degree students in Thailand increased by 9.7 times between 1999 and 2012, from 1,882 to 20,309 students. Most of international students come from neighbor countries[292] like China, Myanmar, Cambodia and Vietnam.[300]
Health

Thailand ranks world's sixth, and Asia's first in the 2019 Global Health Security Index of global health security capabilities in 195 countries,[301] making it the only developing country on the world's top ten. Thailand had 62 hospitals accredited by Joint Commission International.[302] In 2002, Bumrungrad became the first hospital in Asia to meet the standard.[303]
Health and medical care is overseen by the Ministry of Public Health (MOPH), with total national expenditures on health amounting to 4.3 per cent of GDP in 2009. Non-communicable diseases form the major burden of morbidity and mortality, while infectious diseases including malaria[304] and tuberculosis,[305] as well as traffic accidents, are also important public health issues.[306]
In December 2018, the interim parliament voted to legalise the use of cannabis for medical reasons, making Thailand the first Southeast Asian country to allow the use of medical cannabis.[307]
Culture

Thai culture and traditions incorporate influences from India, China, Cambodia, and the rest of Southeast Asia. Thailand's national religion, Theravada Buddhism, is central to modern Thai identity. Thai Buddhism has evolved over time to include many regional beliefs originating from Hinduism, animism, as well as ancestor worship. The official calendar in Thailand is based on the Eastern version of the Buddhist Era (BE). Thai identity today is a social construct of the Phibun regime in the 1940s.[308][309][310]
Several ethnic groups mediated change between their traditional local culture, national Thai, and global cultural influences. Overseas Chinese also form a significant part of Thai society, particularly in and around Bangkok. Thai Chinese businesses prosper as part of the larger bamboo network.[311]

Respect for elderly and superiors (by age, position, monks, or certain professions) is Thai mores, reflecting in many classes of honorifics. Wai is a traditional Thai greeting, and is generally offered first by a person who is younger or lower in social status and position. Older siblings have duties to younger ones.[312]
Taboos in Thai culture include touching someone's head or pointing with the feet, as the head is considered the most sacred and the foot the lowest part of the body.[313]
Art

The origins of Thai art were influenced by Buddhist art and by scenes from the Indian epics. Traditional Thai sculpture almost exclusively depicts images of the Buddha, being very similar with the other styles from Southeast Asia. Traditional Thai paintings usually consist of book illustrations, and painted ornamentation of buildings such as palaces and temples. Thai art was influenced by indigenous civilisations of the Mon and other civilisations. By the Sukothai and Ayutthaya periods, Thai had developed into its own unique style and was later further influenced by the other Asian styles, mostly by Sri Lankan and Chinese. Thai sculpture and painting, and the royal courts provided patronage, erecting temples and other religious shrines as acts of merit or to commemorate important events.[314]
Traditional Thai paintings showed subjects in two dimensions without perspective. The size of each element in the picture reflected its degree of importance. The primary technique of composition is that of apportioning areas: the main elements are isolated from each other by space transformers. This eliminated the intermediate ground, which would otherwise imply perspective. Perspective was introduced only as a result of Western influence in the mid-19th century. Monk artist Khrua In Khong is well known as the first artist to introduce linear perspective to Thai traditional art.[315]
The most frequent narrative subjects for paintings were or are: the Jataka stories, episodes from the life of the Buddha, the Buddhist heavens and hells, themes derived from the Thai versions of the Ramayana and Mahabharata, and scenes of daily life. Some of the scenes are influenced by Thai folklore instead of following strict Buddhist iconography.[314]
Architecture

The Ayutthaya Kingdom movement is designed to display might and riches. The temples in Ayutthaya seldom built eaves stretching from the masterhead.[316] Buddhist temples in Thailand are known as "wats", from the Pāḷi vāṭa, meaning an enclosure: a temple has an enclosing wall that divides it from the secular world. Wat architecture demonstrates many differences in layout and style, but they all adhere to the same principles.[317]
Literature
Thai literature has had a long history. Even before the establishment of the Sukhothai Kingdom there existed oral and written works.[citation needed]
During the Sukhothai Kingdom, most literary works were written in simple prose with certain alliteration schemes. Major works include King Ram Khamhaeng Inscription describing life at the time, which is considered the first literary work in Thai script, but some historians questioned its authenticity.[318] Trai Phum Phra Ruang, written in 1345 by King Maha Thammaracha I, expounds Buddhist philosophy based on an extensive study with reference to over 30 sacred texts and could be considered the nation's first piece of research dissertation.[319]

During the Ayutthaya Kingdom, new poetic forms were created, with different rhyme schemes and metres. It is common to find a combination of different poetic forms in one poetic work. Lilit Yuan Phai is a narrative poem describing the war between King Borommatrailokkanat of Ayutthaya and Prince Tilokaraj of Lan Na. One literary work is Kap He Ruea, composed by Prince Thammathibet in the nirat tradition. Traditionally, the verse is sung during the royal barge procession[320] and has been the model for subsequent poets to emulate. The same prince also composed the greatly admired Kap Ho Khlong on the Visit to Than Thongdaeng and Kap Ho Khlong Nirat Phrabat.[321] The Thonburi period produced Ramakien, a verse drama contributed by King Taksin the Great.
During the 18th century Rattanakosin period, which still fought with the Burmese, many of the early Rattanakosin works dealt with war and military strategy. Some examples are Nirat Rop Phama Thi Tha Din Daeng, Phleng Yao Rop Phama Thi Nakhon Si Thammarat. There were also verse recitals with musical accompaniment, such as Mahori telling the story of Kaki and Sepha, relating the story of Khun Chang Khun Phaen. Other recitals include Sri Thanonchai. The Thai poet Sunthorn Phu is known as "the bard of Rattanakosin" (Thai: กวีเอกแห่งกรุงรัตนโกสินทร์). Sunthorn Phu is best known for his epic poem Phra Aphai Mani, a versified fantasy-adventure novel, a genre of Siamese literature known as nithan kham klon (Thai: นิทานคำกลอน).[321]
Some of the most well-known modern Thai writers include Kukrit Pramoj, Kulap Saipradit, (penname Siburapha), Suweeriya Sirisingh (penname Botan), Chart Korbjitti, Prabda Yoon, and Duanwad Pimwana.[322]
Music and dance

Aside from folk and regional dances (southern Thailand's Menora and Ramwong, for example), the two major forms of Thai classical dance drama are Khon and Lakhon nai. In the beginning, both were exclusively court entertainments, and it was not until much later that a popular style of dance theatre, likay, evolved as a diversion for common folk.[323] Folk dance forms include dance theater forms like likay, numerous regional dances (ram), the ritual dance ram muay, and homage to the teacher, wai khru.[324] Both ram muay and wai khru take place before all traditional muay Thai matches.[citation needed]
The three primary classical ensembles are the Piphat, Khrueang sai, and Mahori. Mahori employ small ching hand cymbals.[325]
Entertainment
Thai films are exported and exhibited in Southeast Asia.[326] Thai cinema has developed its own unique identity.[327] The Thai heist thriller film Bad Genius (2017) was one of the most internationally successful Thai films; it broke Thai film earning records in several Asian countries,[328] Bad Genius won in 12 categories at the 27th Suphannahong National Film Awards, and also won the Jury Award at the 16th New York Asian Film Festival with a worldwide collection of more than $42 million.[329] Shutter (2004) was one of the best-known Thai horror movies and was recognized worldwide.[330] Films such as Ong-Bak: Muay Thai Warrior (2003) and Tom-Yum-Goong (2005), starring Tony Jaa, feature distinctive aspects of Thai martial arts "Muay Thai". Thailand television dramas, known as Lakorn, have become popular in Thailand and regionally.[331]
The entertainment industries are estimated to have directly contributed $2.1 billion in GDP to the Thai economy in 2011. They also directly supported 86,600 jobs.[332] Amongst several dance-pop artists who have made internationally successful are "Lisa" Lalisa Manobal,[333] Violette Wautier,[334] and Tata Young.
Cuisine

.jpg)


Thai cuisine is one of the most popular in the world.[335] Common ingredients include garlic, lemongrass, kaffir lime, galangal, turmeric, coriander, and coconut milk.[336][337][338][339][340] Each region of Thailand has its specialities: kaeng khiao wan (green curry) in the central region, som tam (green papaya salad) in the northeast, khao soi in the north, and massaman curry in the south.[citation needed]
In 2017, seven Thai dishes appeared on a list of the "World's 50 Best Foods"—an online worldwide poll by CNN Travel. Thailand had more dishes on the list than any other country. They were: tom yam goong (4th), pad Thai (5th), som tam (6th), massaman curry (10th), green curry (19th), Thai fried rice (24th) and nam tok mu (36th).[341] Two desserts were also listed in CNN's 50 Best Desserts Around The World: mango sticky rice and tub tim krob.[342]
The staple food in Thailand is rice, particularly jasmine rice, which forms part of almost every meal. Thailand is a leading exporter of rice, and Thais consume over 100 kg of milled rice per person per year.[343] Thailand is also the world leader in edible insect industry[344] and well known for its street food; Bangkok is sometimes called the street food capital of the world.[345][346]
Units of measurement
Thailand generally uses the metric system, but traditional units of measurement for land area are used, and imperial units of measurement are occasionally used for building materials. Years are numbered as B.E. (Buddhist Era) in educational settings, civil service, government, contracts, and newspaper datelines. However, in banking, and increasingly in industry and commerce, standard Western year (Christian or Common Era) counting is the standard practice.[347]
Sports
.jpeg)
Muay Thai (lit. 'Thai boxing') is a combat sport that uses stand-up striking along with various clinching techniques. Muay Thai became widespread internationally in the late-20th to 21st centuries. Famous practitioners include Buakaw Banchamek, Samart Payakaroon, and Apidej Sit-Hirun.[348] Association football has overtaken Muay Thai as the most widely followed sport in Thailand. The Thailand national football team has played the AFC Asian Cup six times and reached the semifinals in 1972. The country has hosted the Asian Cup twice, in 1972[349] and in 2007 (along with Indonesia, Malaysia, and Vietnam for the 2007).
Volleyball is rapidly growing as one of the most popular sports. The women's team has often participated in the World Championship, World Cup, and World Grand Prix Asian Championship. They have won the Asian Championship twice and the Asian Cup once. Takraw is a sport native to Thailand in which the players hit a rattan ball and are only allowed to use their feet, knees, chest, and head to touch the ball. Sepak takraw is a form of this sport which is similar to volleyball. A rather similar game but played only with the feet is buka ball.
Rugby is also a growing sport in Thailand with the Thailand national rugby union team rising to be ranked 61st in the world.[350] Thailand became the first country in the world to host an international 80 welterweight rugby tournament in 2005.[351] Thailand has also attracts golfers from Japan, Korea, and Western countries.[352] There are more than 200 world-class golf courses nationwide.[353] For basketball, the Chang Thailand Slammers won the 2011 ASEAN Basketball League Championship.[354] The Thailand national basketball team had its most successful year at the 1966 Asian Games where it won the silver medal.[355]

The Lumpinee Boxing Stadium originally sited at Rama IV Road near Lumphini Park hosted its final Muay Thai boxing matches on 8 February 2014 after the venue first opened in December 1956. On 11 February 2014, the stadium was relocated to Ram Intra Road due to the new venue's capacity.[356] Thammasat Stadium in Bangkok was built for the 1998 Asian Games. Rajamangala National Stadium is the biggest sporting arena in Thailand, with a capacity of around 50,000.[357]
See also
Notes
- ^ a b /ˈtaɪlænd, -lənd/, TYE-land, -lənd; Thai: ประเทศไทย, RTGS: Prathet Thai, pronounced [pratʰêːt tʰaj]
- ^ a b /saɪˈæm, ˈsaɪæm/, sye-AM, SYE-am; Thai: สยาม, RTGS: sayam, pronounced [sajǎːm]; also spelled Siem, Syâm, or Syâma
- ^ Through the following chain: *kəri: > *kəli: > *kədi:/*kədaj > *di:/*daj > *dajA (Proto-Southwestern Tai) > tʰajA2 (in Siamese and Lao) or > tajA2 (in the other Southwestern and Central Tai languages classified by Li Fangkuei).[17] Ferlus work is based on simple rules of phonetic change observable in the Sinosphere and studied for the most part by William H. Baxter (1992).
- ^ "Ayutthaya emerged as a dominant centre in the late 14th century. The Chinese called this region Xian, which the Portuguese converted into Siam."
- ^ See #Ethnic groups section.
- ^ The 2016 Thai constitutional referendum was held on 7 August 2016. Its ratification was held on 6 April 2017.[112]
- ^ Such as: "constitutional dictatorship" or "parliamentary dictatorship,"[138] " military coup regime,"[139] "semicivilian" or "semi-elected,"[140] "managed democracy,"[141] and "guided democracy."[142]
References
- ^ a b c d e f g "Thailand" Archived 10 June 2021 at the Wayback Machine, The World Factbook.
- ^ a b c d "Population by religion, region and area, 2018". NSO. Archived from the original on 24 April 2021. Retrieved 9 March 2021.
- ^ "Population statistics of the civil registration (monthly)".
- ^ National Statistics Office, "100th anniversary of population censuses in Thailand: Population and housing census 2010: 11th census of Thailand". (in Thai) Archived 12 July 2012 at the Wayback Machine. popcensus.nso.go.th.
- ^ a b c d "World Economic Outlook Database, April 2024 Edition. (Thailand)". imf.org. International Monetary Fund. 16 April 2024. Archived from the original on 16 April 2024. Retrieved 16 April 2024.
- ^ "Gini Index". World Bank. Archived from the original on 27 July 2018. Retrieved 12 August 2021.
- ^ "Human Development Report 2023/2024" (PDF). United Nations Development Programme. 13 March 2024. Archived (PDF) from the original on 13 March 2024. Retrieved 13 March 2024.
- ^ "Multiple Factors Behind Thailand's Birth Rate Decline, Experts Say". Voice of America. 23 March 2024. Retrieved 6 July 2024.
- ^ "Land Area". The Government Public Relations Department. Retrieved 1 June 2024.
- ^ "Capital City". The Government Public Relations Department. Retrieved 1 June 2024.
- ^ Nuchkoom Smith, Nucharee; Smith, Robert Brian (1 October 2019). "Has Thailand learnt any Lessons from the Bowring Treaty and the Treaty of Amity?" (PDF). Athens Journal of Law. 5 (4): 405–418. doi:10.30958/ajl.5-4-3. ISSN 2407-9685. S2CID 211453326. Archived (PDF) from the original on 11 May 2022. Retrieved 17 April 2022.
- ^ *Abuza, Zachary (27 September 2021). "Thailand's Constitutional Dictatorship Weathers the Storm". The Diplomat. Retrieved 14 March 2022.
- Glassman, Jim (2020). "Lineages of the Authoritarian State in Thailand: Military Dictatorship, Lazy Capitalism and the Cold War Past as Post-Cold War Prologue". Journal of Contemporary Asia. 50 (4): 571–592. doi:10.1080/00472336.2019.1688378. S2CID 211436855.
- Bandow, Doug (3 December 2020). "Thailand's Military Is Getting Ready for Another Crackdown". Foreign Policy. Retrieved 14 March 2022.
- ^ Thailand and the World Bank Archived 16 December 2005 at the Wayback Machine, World Bank on Thailand country overview.
- ^ The Guardian, Country profile: Thailand Archived 23 May 2010 at the Wayback Machine, 25 April 2009.
- ^ a b Cœdès, George (1968). Walter F. Vella (ed.). The Indianized States of Southeast Asia. Trans. Susan Brown Cowing. University of Hawaii Press. ISBN 978-0-8248-0368-1.
- ^ a b c Phumisak, Chit (1992). ความเป็นมาของคําสยาม ไทย, ลาว และขอม และลักษณะทางสังคมของชื่อชนชาติ: ฉบับสมบูรณ์ เพิ่มเติม ข้อเท็จจริงว่าด้วยชนชาติขอม [Etymology of Siam, Thai, Lao, Khmer] (in Thai). Samnakphim Sayām. ISBN 978-974-85729-9-4. Archived from the original on 28 March 2024. Retrieved 31 December 2021.
- ^ Ferlus, Michel (2009). Formation of Ethnonyms in Southeast Asia Archived 19 November 2016 at the Wayback Machine. 42nd International Conference on Sino-Tibetan Languages and Linguistics, November 2009, Chiang Mai, 2009, p.3.
- ^ Pain, Frédéric (2008). "An Introduction to Thai Ethnonymy: Examples from Shan and Northern Thai". Journal of the American Oriental Society. 128 (4): 641–662. JSTOR 25608449.
- ^ Panyasuppakun, Kornrawee (8 August 2017). "Patriotism remixed". Bangkok Post. Retrieved 1 June 2024.
- ^ Barend Jan Terwiel, Chaichuen Khamdaengyodtai, Shan Manuscripts. Franz Steiner, 2003, p. 9.
- ^ Eliot, Charles (1921). The Project Gutenberg EBook of Hinduism and Buddhism, An Historical Sketch, Vol. 3 (of 3) [EBook #16847]. London: Routledge & Kegan Paul Ltd. pp. Ch. xxxvii 1, citing in turn Footnote 189: The name is found on Champan inscriptions of 1050 CE and according to Gerini appears in Ptolemy's Samarade = Sâmaraṭṭha. See Gerini, Ptolemy, p. 170. But Samarade is near Bangkok and there can hardly have been Thais there in Ptolemy's time, and Footnote 190: So too in Central Asia Kustana appears to be a learned distortion of the name Khotan, made to give it a meaning in Sanskrit.
- ^ Klikauer, Thomas (2008), Klikauer, Thomas (ed.), "Distorted Communication I: Classifications", Management Communication: Communicative Ethics and Action, London: Palgrave Macmillan UK, pp. 55–73, doi:10.1057/9780230583238_4, ISBN 978-0-230-58323-8, archived from the original on 28 March 2024, retrieved 2 January 2024
- ^ a b c d Baker, Christopher; Phongpaichit, Pasuk (2014). A History of Thailand. Singapore: C.O.S Printers Pte Ltd. ISBN 978-1-107-42021-2.
- ^ "จารึกวัดศรีชุม" [Wat Sri Chum Inscription] (in Thai). Princess Maha Chakri Sirindhorn Anthropology Centre. Archived from the original on 28 August 2023. Retrieved 29 August 2023.
- ^ Thailand (Siam) History, CSMngt-Thai. Archived 24 April 2015 at the Wayback Machine
- ^ a b c d e f g Barbara Leitch LePoer (1989). Thailand: A Country Study. Federal Research Division, Library of Congress.
- ^ a b c d e Baker, Chris; Phongpaichit, Pasuk (2017). A History of Ayutthaya. Cambridge University Press. ISBN 978-1-107-19076-4. Archived from the original on 28 March 2024. Retrieved 15 December 2020.
- ^ Tsang, Cheng-hwa (24 January 2008). "Recent advances in the Iron Age archaeology of Taiwan". Bulletin of the Indo-Pacific Prehistory Association. 20. doi:10.7152/bippa.v20i0.11751 (inactive 5 August 2024). ISSN 1835-1794.
{{cite journal}}: CS1 maint: DOI inactive as of August 2024 (link) - ^ Turton, M. (2021). Notes from central Taiwan: Our brother to the south. Taiwan's relations with the Philippines date back millennia, so it's a mystery that it's not the jewel in the crown of the New Southbound Policy. Taiwan Times.
- ^ Everington, K. (2017). Birthplace of Austronesians is Taiwan, capital was Taitung: Scholar. Taiwan News.
- ^ Bellwood, P., H. Hung, H., Lizuka, Y. (2011). Taiwan Jade in the Philippines: 3,000 Years of Trade and Long-distance Interaction. Semantic Scholar.
- ^ Higham, Charles; Higham, Thomas; Ciarla, Roberto; Douka, Katerina; Kijngam, Amphan; Rispoli, Fiorella (10 December 2011). "The Origins of the Bronze Age of Southeast Asia". Journal of World Prehistory. 24 (4): 227–274. doi:10.1007/s10963-011-9054-6. S2CID 162300712. Archived from the original on 11 May 2021. Retrieved 10 February 2018 – via Researchgate.net.
- ^ Thailand. History Archived 2 April 2012 at the Wayback Machine. Encyclopædia Britannica
- ^ a b c d e f g Wyatt, David K. (1984). Thailand: A Short History. New Haven: Yale University Press. ISBN 978-0-300-03054-9.
- ^ E. Jane Keyes; James A. Hafner; et al. (2018). "Thailand: History". Encyclopædia Britannica. Archived from the original on 24 June 2021. Retrieved 4 April 2018.
- ^ Keyes, Charles F. (1997). "Cultural Diversity and National Identity in Thailand". In Michael E. Brown; Sumit Ganguly (eds.). Government policies and ethnic relations in Asia and the Pacific. MIT Press. pp. 197–232. ISBN 9780262522458.
- ^ a b c Kutanan, Wibhu; Liu, Dang; Kampuansai, Jatupol; Srikummool, Metawee; Srithawong, Suparat; Shoocongdej, Rasmi; Sangkhano, Sukrit; Ruangchai, Sukhum; Pittayaporn, Pittayawat; Arias, Leonardo; Stoneking, Mark (2021). "Reconstructing the Human Genetic History of Mainland Southeast Asia: Insights from Genome-Wide Data from Thailand and Laos". Mol Biol Evol. 38 (8): 3459–3477. doi:10.1093/molbev/msab124. PMC 8321548. PMID 33905512.
- ^ a b Wibhu Kutanan; Jatupol Kampuansai; Metawee Srikummool; Daoroong Kangwanpong; Silvia Ghirotto; Andrea Brunelli; Mark Stoneking (2016). "Complete mitochondrial genomes of Thai and Lao populations indicate an ancient origin of Austroasiatic groups and demic diffusion in the spread of Tai-Kadai languages". Human Genetics. 136 (1): 85–98. doi:10.1007/s00439-016-1742-y. hdl:11858/00-001M-0000-002C-0639-D. PMC 5214972. PMID 27837350. Archived from the original on 18 January 2024.
- ^ a b Wibhu Kutanan; Jatupol Kampuansai; Andrea Brunelli; Silvia Ghirotto; Pittayawat Pittayaporn; Sukhum Ruangchai; Roland Schröder; Enrico Macholdt; Metawee Srikummool; Daoroong Kangwanpong; Alexander Hübner; Leonardo Arias Alvis; Mark Stoneking (2017). "New insights from Thailand into the maternal genetic history of Mainland Southeast Asia". European Journal of Human Genetics. 26 (6): 898–911. doi:10.1038/s41431-018-0113-7. hdl:21.11116/0000-0001-7EEF-6. PMC 5974021. PMID 29483671. Archived from the original on 18 January 2024. Retrieved 19 January 2024.
- ^ "ชนชาติไทย 'ไม่ใช่' คนไทย โดย สุจิตต์ วงษ์เทศ". Matichon. Archived from the original on 20 February 2024. Retrieved 20 February 2024.
- ^ a b c Smith, John (2019). State, Community, and Ethnicity in Early Modern Thailand, 1351–1767 (PhD thesis). University of Michigan. hdl:2027.42/151629. Archived from the original on 28 March 2024. Retrieved 5 April 2023.
- ^ Juntanamalaga, Preecha (1 June 1988). "Thai or Siam?". Names. 36 (1): 69–84. doi:10.1179/nam.1988.36.1-2.69. ISSN 1756-2279. Archived from the original on 28 March 2024. Retrieved 5 April 2023.
- ^ Chamberlain, James R. (2016). "Kra-Dai and the Proto-History of South China and Vietnam". Journal of the Siam Society. 104: 27–77. Archived from the original on 3 January 2023. Retrieved 18 January 2024.
- ^ Baker, Chris (2002), "From Yue To Tai" (PDF), Journal of the Siam Society, 90 (1–2): 1–26, archived (PDF) from the original on 4 March 2016, retrieved 3 May 2018
- ^ Taylor, Keith W. (1991), The Birth of Vietnam, University of California Press, ISBN 978-0-520-07417-0, archived from the original on 7 July 2023, retrieved 1 November 2020
- ^ Baker & Phongpaichit 2017, p. 26.
- ^ Evans, Grant (2002), A Short History of Laos: The Land in Between (PDF), Allen & Unwin, ISBN 978-1-86448-997-2, archived (PDF) from the original on 29 February 2024, retrieved 18 January 2024.
- ^ Du, Yuting; Chen, Lufan (1989). "Did Kublai Khan's Conquest of the Dali Kingdom Give Rise to the Mass Migration of the Thai People to the South?" (PDF). Journal of the Siam Society. 77 (1c). Archived (PDF) from the original on 9 September 2020. Retrieved 18 January 2024.
The Thai people in the north as well as in the south did not in any sense "migrate en masse to the south" after Kublai Khan's conquest of the Dali Kingdom
- ^ Jumsai, M.L. Manich (5 August 1967). History of Laos. Chalermnit. ISBN 978-974-7390-21-6.
- ^ Ratanavongsa, Prince Phetsarath (1978). The Iron Man of Laos. Dalley Book Service, Inc.
- ^ a b Pittayaporn, Pittayawat (2014). Layers of Chinese Loanwords in Proto-Southwestern Tai as Evidence for the Dating of the Spread of Southwestern Tai Archived 27 June 2015 at the Wayback Machine. MANUSYA: Journal of Humanities, Special Issue No 20: 47–64.
- ^ a b "Wiang Nong Lom Cultural Heritage" (PDF). Fine Arts Department of Thailand. 2023. Archived from the original (PDF) on 21 January 2024. Retrieved 21 January 2024.
- ^ a b "เมืองโยนก นครในตำนานล้านนา" (in Thai). Fine Arts Department of Thailand. 2022. Archived from the original on 18 January 2024. Retrieved 18 January 2024.
- ^ พิเศษ เจียจันทร์พงษ์ (28 January 2022). "พระเจ้าพรหมมหาราช ในตำนานล้านนา นัยสำคัญของกษัตริย์สืบสายทางธรรม VS สายเลือด". silpa-mag.com (in Thai). Archived from the original on 23 January 2024. Retrieved 23 January 2024.
- ^ a b ""รอยเลื่อนแม่จัน" กับ "โยนกเชียงแสน" ตำนานที่อาจมีอายุมากกว่า 1,800 ปี". Manager Daily (in Thai). 24 April 2020. Archived from the original on 18 January 2024.
- ^ Du & Chen (1989), p. 38
- ^ Hou Hanshu vol. 5 Archived 8 October 2023 at the Wayback Machine "九年春正月, 永昌徼外蠻夷及撣國重譯奉貢."
- ^ Hou Hanshu vol. 6 Archived 8 October 2023 at the Wayback Machine txt: "十二月, 永昌徼外撣國遣使貢獻."
- ^ Hou Hanshu vol. 7 Archived 8 October 2023 at the Wayback Machine txt: "十二月, 日南徼外葉調國、撣國遣使貢獻."
- ^ Maha Sila Viravond. "HISTORY OF LAOS" (PDF). Refugee Educators' Network. Archived from the original (PDF) on 3 April 2020. Retrieved 29 December 2017.
- ^ M.L. Manich. "HISTORY OF LAOS (includlng the hlstory of Lonnathai, Chiangmai)" (PDF). Refugee Educators' Network. Archived from the original (PDF) on 8 October 2021. Retrieved 29 December 2017.
- ^ Wood, Spencer H.; Wood, Layle R.; Ziegler, Alan D. (2 November 2015). "Natural degradation of earthworks, trenches, walls and moats, Northern Thailand". Journal of Field Archaeology. 40 (6): 675–694. doi:10.1080/00934690.2015.1103645. ISSN 0093-4690. S2CID 32414373. Archived from the original on 18 January 2024. Retrieved 18 January 2024.
- ^ "ตามหาเมืองเงินยาง ตอน 3". finearts.go.th (in Thai). Archived from the original on 3 November 2023. Retrieved 10 November 2023.
- ^ Coedès, George (1968). Walter F. Vella (ed.). The Indianized States of Southeast Asia. trans.Susan Brown Cowing. University of Hawaii Press. ISBN 978-0-8248-0368-1.
- ^ a b Thepthani, Phra Borihan (1953). Thai National Chronicles: the history of the nation since ancient times (in Thai). S. Thammasamakkhi. Archived from the original on 5 November 2023. Retrieved 5 November 2023.
- ^ a b c สงบ สุริยินทร์ (30 December 2022). "เมืองลพบุรีเป็นของไทยเมื่อใด?". silpa-mag.com (in Thai). Archived from the original on 25 December 2023. Retrieved 25 December 2023.
- ^ โรม บุนนาค (7 April 2023). "๒ มหาราชของคนไทยก่อนเกิดประเทศไทย! ๒ เมืองหลวงเป็นอำเภอและจังหวัดในปัจจุบัน!!". Manager Daily (in Thai). Archived from the original on 19 January 2024. Retrieved 19 January 2024.
- ^ "ประวัติของจังหวัดเชียงราย" [History of Chiang Rai Province] (in Thai). Chiang Rai Provincial Administrative Organization. 2024. Archived from the original on 21 January 2024. Retrieved 21 January 2024.
- ^ a b องค์ บรรจุน (10 December 2022). "ค้นหาร่องรอยภาษามอญ ในภาคอีสานของไทย". silpa-mag.com (in Thai). Archived from the original on 16 January 2024. Retrieved 17 January 2024.
- ^ สุจิตต์ วงษ์เทศ (9 August 2018). "สุจิตต์ วงษ์เทศ : ชาวนอกอยู่ภาคใต้ คนเมืองในอยู่ภาคกลาง" (in Thai). Matichon. Archived from the original on 26 January 2024. Retrieved 26 January 2024.
- ^ Baker, Christopher (2014). A history of Thailand. Melbourne, Australia: Cambridge University Press. pp. 3–4. ISBN 978-1-316-00733-4.
- ^ พระรัตนปัญญาเถระ (1958). "ชินกาลมาลีปกรณ์" (in Thai). Fine Arts Department of Thailand. Archived from the original on 28 March 2024. Retrieved 17 January 2024.
- ^ "จาก "เสียม (สยาม)" สู่ "ไถ (ไทย)": บริบทและความหมายในการรับรู้ของชาวกัมพูชา". silpa-mag.com (in Thai). March 2009. Archived from the original on 23 December 2023. Retrieved 23 December 2023.
- ^ a b "เส้นทางศรีวิชัย : เครือข่ายทางการค้าที่ยิ่งใหญ่ที่สุดในทะเลใต้ยุคโบราณ ตอน ราชวงศ์ไศเลนทร์ที่จัมบิ (ประมาณ พ.ศ.1395–1533) (ตอนจบ)". Manager Daily (in Thai). 1 December 2023. Archived from the original on 23 December 2023. Retrieved 23 December 2023.
- ^ [1] Archived 28 August 2009 at the Wayback Machine
- ^ สุจิตต์ วงษ์เทศ (4 December 2023). "คนโคราช ไม่ใช่ "ลาว" แล้วคนโคราชเป็นใคร? มาจากไหน?". silpa-mag.com (in Thai). Archived from the original on 18 January 2024. Retrieved 19 January 2024.
- ^ Wyatt, David K. (2003). Thailand: A Short History. Yale University Press. ISBN 978-0-300-08475-7.
- ^ เพ็ญสุภา สุขคตะ (17 November 2022). "พระนางจามเทวี จารึกศรีวิชัย สายสัมพันธ์ขอมเจนละ-จามปา ในมุมมองของ ผศ.พงศ์เกษม สนธิไทย" (in Thai). Matichon. Archived from the original on 16 January 2024. Retrieved 17 January 2024.
- ^ "Corpus of the Inscriptions of Campā: C. 3 Lintel from Phan Rang". New York University. Archived from the original on 16 January 2024. Retrieved 17 January 2024.
- ^ Higham, Charles (1989). The Archaeology of Mainland Southeast Asia. Cambridge University Press. ISBN 0-521-27525-3. Archived from the original on 28 March 2024. Retrieved 6 September 2009.
- ^ เกษตรศิริ, ชาญวิทย์ (2005). อยุธยา: ประวัติศาสตร์และการเมือง. โรงพิมพ์มหาวิทยาลัยธรรมศาสตร์. ISBN 978-974-91572-7-5.
- ^ a b c d e f g h i j k l m n o p q r s t u v w x y z aa ab ac ad ae af ag ah ai aj ak al am Wyatt, David K. (2013). Thailand: A Short History [ประวัติศาสตร์ไทยฉบับสังเขป] (in Thai). Translated by ละอองศรี, กาญจนี. มูลนิธิโครงการตำราสังคมศาสตร์และมนุษยศาสตร์, มูลนิธิโตโยต้าประเทศไทย. ISBN 978-616-7202-38-9.
- ^ "Ayutthaya history Foreign Settlements". Archived from the original on 6 July 2020. Retrieved 24 May 2020.
- ^ Harvey, G E (1925). History of Burma. London: Frank Cass & Co. Ltd.
- ^ Ruangsilp, Bhawan (2007). Dutch East India Company Merchants at the Court of Ayutthaya: Dutch Perceptions of the Thai Kingdom c. 1604–1765. Leiden, Netherlands: Koninklijke Brill NV. ISBN 978-0-300-08475-7. Archived from the original on 28 June 2023. Retrieved 20 November 2009.
- ^ จรรยา ประชิตโรมรัน. (2548). สมเด็จพระเจ้าตากสินมหาราช. สำนักพิมพ์แห่งจุฬาลงกรณ์มหาวิทยาลัย. หน้า 55
- ^ Nolan, Cathal J. (2002). The Greenwood Encyclopedia of International Relations: S-Z by Cathal J. Nolan. Greenwood Publishing. ISBN 978-0-313-32383-6. Archived from the original on 28 March 2024. Retrieved 21 November 2015.
- ^ Hwa, Cheng Siok (1971). "The Crawford Papers – A Collection of Official Records relating to the Mission of Dr. John Crawfurd sent to Siam by the Government of India in the year 1821". Journal of Southeast Asian Studies. 3 (2): 324–325. doi:10.1017/S0022463400019421 (inactive 26 August 2024).
{{cite journal}}: CS1 maint: DOI inactive as of August 2024 (link) - ^ "Ode to Friendship, Celebrating Singapore – Thailand Relations: Introduction". National Archives of Singapore. 2004. Archived from the original on 3 March 2007. Retrieved 24 April 2007.
- ^ "King, country and the coup". The Indian Express. Mumbai. 22 September 2006. Archived from the original on 14 May 2011. Retrieved 3 November 2011.
- ^ Declaration between Great Britain and France with regard of the Kingdom of Siam and other matters Archived 31 March 2017 at the Wayback Machine London. 15 January 1896. Treaty Series. No. 5
- ^ Werner Gruhl, Imperial Japan's World War Two, 1931–1945 Archived 28 March 2024 at the Wayback Machine, Transaction Publishers, 2007 ISBN 978-0-7658-0352-8
- ^ Fine, Herbert A. (1965). "The Liquidation of World War II in Thailand". Pacific Historical Review. 34 (1): 65–82. doi:10.2307/3636740. ISSN 0030-8684. JSTOR 3636740.
- ^ a b "The 1973 revolution and its aftermath". Encyclopædia Britannica. Archived from the original on 11 April 2019. Retrieved 23 August 2019.
- ^ "Thailand ..Communists Surrender En Masse". Ottawa Citizen. 2 December 1982. Archived from the original on 1 April 2020. Retrieved 8 August 2023.
- ^ "Partial democracy and the search for a new political order". Encyclopædia Britannica. Archived from the original on 23 March 2018. Retrieved 11 March 2018.
- ^ "Asw". Human Rights Watch. Archived from the original on 10 December 2022. Retrieved 8 August 2023.
- ^ Thailand: The massacre in Bangkok (PDF) (Report). Amnesty International. October 1992. Archived (PDF) from the original on 10 August 2023. Retrieved 8 August 2023.
- ^ Warr, Peter (2007). Thailand Beyond the Crisis. Routledge Curzon. ISBN 978-1-134-54151-5.
- ^ "Thailand Letter of Intent, November 25, 1997". imf.org. Archived from the original on 2 January 2024. Retrieved 2 January 2024.
- ^ "Concerns arise over warning systems as Boxing Day marks 19 years since 2004 tsunami". nationthailand. 26 December 2023. Retrieved 19 May 2024.
- ^ "Thailand Economic Monitor, November 2005" (PDF). Archived from the original (PDF) on 2 September 2009. Retrieved 19 February 2010.
- ^ Na Ranong, Viroj, Na Ranong, Anchana, Universal Health Care Coverage: Impacts of the 30-Baht Health Care Scheme on the Rural Poor in Thailand, TDRI Quarterly Review, September 2006
- ^ a b Phongpaichit, Pasuk (December 2008). "Thailand: Fighting Over Democracy". Economic and Political Weekly. 43 (50): 18–21.
- ^ Connors, Michael K. (28 November 2008). "Thailand-Four elections and a coup". Australian Journal of International Affairs. 62 (4): 478, 483–484. doi:10.1080/10357710802480717. ISSN 1035-7718. S2CID 154415628. Archived from the original on 11 August 2023. Retrieved 8 August 2023.
- ^ Erawan EMS Center, รายชื่อผู้เสียชีวิตจากสถานการณ์การชุมนุมของกลุมนปช. Archived 6 March 2012 at the Wayback Machine
- ^ "PDRC leaders jailed for terrorism, insurrection over street rallies". Bangkok Post. Archived from the original on 3 January 2024. Retrieved 3 January 2024.
- ^ "Protests as Thailand senators debate amnesty bill". The Guardian. 11 November 2013. Archived from the original on 3 December 2013. Retrieved 10 April 2019.
- ^ Prasirtsuk, Kitti (2015). "Thailand in 2014: Another Coup, a Different Coup?". Asian Survey. 55 (1): 200–206. doi:10.1525/as.2015.55.1.200. ISSN 0004-4687. JSTOR 10.1525/as.2015.55.1.200. Retrieved 3 June 2024.
- ^ a b Beech, Hannah (8 February 2019). "Thailand's King Rejects His Sister's Candidacy for Prime Minister". The New York Times. ISSN 0362-4331. Archived from the original on 13 February 2019. Retrieved 11 February 2019.
- ^ Taylor, James (1 September 2021). "Thailand's new right, social cleansing and the continuing military–monarchy entente". Asian Journal of Comparative Politics. 6 (3): 253–273. doi:10.1177/2057891120980835. ISSN 2057-8911. S2CID 234182253. Archived from the original on 18 November 2021. Retrieved 18 November 2021.
- ^ Thai King Signs Military-Backed Constitution Archived 10 April 2019 at the Wayback Machine, NPR, 6 April 2017
- ^ Montesano, Michael J. (2019). "The Place of the Provinces in Thailand's Twenty-Year National Strategy: Toward Community Democracy in a Commercial Nation?" (PDF). ISEAS Perspective. 2019 (60): 1–11. Archived (PDF) from the original on 13 September 2020. Retrieved 23 August 2020.
- ^ "Thailand election results delayed as allegations of cheating grow". Australia: ABC News. 25 March 2019. Archived from the original on 26 March 2019. Retrieved 26 March 2019.
- ^ "Thai protesters stage biggest anti-government demonstration in years". France 24. 16 August 2020. Archived from the original on 23 September 2020. Retrieved 20 September 2020.
- ^ "Thailand: youthful protesters break the kingdom's biggest political taboo". Financial Times. London. 27 August 2020. Archived from the original on 10 December 2022.
- ^ "[Full statement] The demonstration at Thammasat proposes monarchy reform". Prachatai English. 11 August 2020. Archived from the original on 20 August 2020. Retrieved 23 August 2020.
- ^ Cunningham, Philip J. "An unexpectedly successful protest". Bangkok Post. Archived from the original on 8 November 2020. Retrieved 24 September 2020.
- ^ Rasheed, Zaheena. "'Impressive victory': Thai opposition crushes military parties". Al Jazeera. Archived from the original on 25 August 2023. Retrieved 1 September 2023.
- ^ "Srettha Thavisin elected Thailand PM as Thaksin returns from exile". Al Jazeera. Archived from the original on 1 September 2023. Retrieved 1 September 2023.
- ^ Wongcha-um, Panu; Setboonsarng, Chayut (14 August 2024). "Thai court dismisses PM Srettha over cabinet appointment". Reuters. Retrieved 14 August 2024.
- ^ a b c d e f g h i j k l m "The Climate of Thailand" (PDF). Thai Meteorological Department. Archived from the original (PDF) on 1 August 2016. Retrieved 18 August 2016.
- ^ Dr. Susan L. Woodward (1997–2014). "Tropical Savannas". Biomes of the World. S. L. Woodward. Archived from the original on 19 December 2013. Retrieved 23 February 2014.
- ^ Overland, Indra et al. (2017) Impact of Climate Change on ASEAN International Affairs: Risk and Opportunity Multiplier Archived 28 July 2020 at the Wayback Machine, Norwegian Institute of International Affairs (NUPI) and Myanmar Institute of International and Strategic Studies (MISIS).
- ^ "Report: Flooded Future: Global vulnerability to sea level rise worse than previously understood". climatecentral.org. 29 October 2019. Archived from the original on 4 September 2022. Retrieved 5 September 2022.
- ^ a b "Thailand's Elephants". Thai Elephant Conservation Center. Archived from the original on 5 March 2015. Retrieved 3 March 2015.
- ^ "Five New National Parks in Thailand". The Government Public Relations Department. 6 August 2019. Retrieved 4 January 2022.[permanent dead link]
- ^ "2016 Report". EPI Report. Archived from the original on 4 February 2016. Retrieved 17 December 2016.
- ^ EPI (2016): Thailand Archived 27 December 2016 at the Wayback Machine
- ^ Grantham, H. S.; et al. (2020). "Anthropogenic modification of forests means only 40% of remaining forests have high ecosystem integrity – Supplementary Material". Nature Communications. 11 (1): 5978. Bibcode:2020NatCo..11.5978G. doi:10.1038/s41467-020-19493-3. ISSN 2041-1723. PMC 7723057. PMID 33293507.
- ^ "Poaching for meat poses new extinction risk to Thai elephants". The Guardian. Associated Press. 26 January 2012. Archived from the original on 5 February 2018. Retrieved 4 February 2018.
- ^ Hile, Jennifer (6 October 2002). "Activists Denounce Thailand's Elephant "Crushing" Ritual". National Geographic Today. Archived from the original on 18 February 2007. Retrieved 7 June 2007.
- ^ Stiles, Daniel. The Elephant and Ivory Trade in Thailand (PDF) (Report). Traffic Southeast Asia. pp. 1–2.
- ^ Teena Amrit Gill (18 February 1997). "Endangered Animals on Restaurant Menus". Albion Monitor/News. Archived from the original on 16 May 2007. Retrieved 7 June 2007.
- ^ "Thai Forests: Dept. National Parks, Wildlife & Plants". Thai Society for the Conservation of Wild Animals. Archived from the original on 26 November 2014.
- ^ "A list of previous coups in Thailand". Associated Press. 19 September 2006. Archived from the original on 16 October 2007. Retrieved 25 April 2010.
- ^ "Raw Data: List of Recent Coups in Thailand's History". Fox News. 19 September 2006. Archived from the original on 6 July 2008. Retrieved 25 April 2010.
- ^ Abuza, Zachary (27 September 2021). "Thailand's Constitutional Dictatorship Weathers the Storm". The Diplomat. Retrieved 14 March 2022.
- ^ Glassman, Jim (2020). "Lineages of the Authoritarian State in Thailand: Military Dictatorship, Lazy Capitalism and the Cold War Past as Post-Cold War Prologue". Journal of Contemporary Asia. 50 (4): 571–592. doi:10.1080/00472336.2019.1688378. Retrieved 14 March 2022.
- ^ "Thailand: Freedom in the World 2021 Country Report". Freedom House. Retrieved 28 April 2022.
- ^ "Prem Tinsulanonda's Legacy—and the Failures of Thai Politics Today". Council on Foreign Relations. Retrieved 28 April 2022.
- ^ "Election observers call still-partial Thai vote count flawed". AP NEWS. 27 March 2019. Retrieved 28 April 2022.
- ^ สถิติที่ไม่น่าภูมิใจเมื่อไทยติดอันดับที่ 4 ประเทศที่มีการรัฐประหารบ่อยที่สุดในโลก. Siam Intelligence (in Thai). Archived from the original on 6 April 2018. Retrieved 6 April 2018.
- ^ Gray, Denis D. (22 August 2015). "Deadly bombing in military-ruled Thailand adds to mounting woes in one-time 'Land of Smiles'". U.S. News & World Report. Associated Press. Archived from the original on 22 August 2015. Retrieved 23 August 2015.
- ^ "A guide to Thailand's next Senate and 'the most complicated election in the world'". 17 April 2024. Retrieved 22 April 2024.
- ^ ""มงคล สุระสัจจะ"ผงาดนั่ง "ประธานวุฒิสภา" คนใหม่ ด้วยมติสว.ท่วมท้น 159 คะแนน". thansettakij (in Thai). 23 July 2024. Retrieved 23 July 2024.
- ^ "Thailand's juristocracy". 17 May 2014. Archived from the original on 5 September 2015.
- ^ Teehankee, Julio; Tiulegenov, Medet; Wang, Yi-ting; Ciobanu, Vlad; Lindberg, Staffan I. "Party System in South and Southeast Asia: A Thematic Report Based on Data 1900–2012". V-Dem Thematic Report Series, No. 2, October 2013.
- ^ a b Croissant, Aurel; Völkel, Philip (21 December 2010). "Party system types and party system institutionalization: Comparing new democracies in East and Southeast Asia". Party Politics. 18 (2). doi:10.1177/1354068810380096. S2CID 145074799.
- ^ McCargo, Duncan, "Network monarchy and legitimacy crises in Thailand", The Pacific Review, volume 18, issue 4, December 2005
- ^ Head, Jonathan (5 December 2007). "Why Thailand's king is so revered". BBC News. Archived from the original on 17 February 2009. Retrieved 17 October 2015.
- ^ Denby, Kenneth. "Thai protests: The king who made himself a gift to republicans". The Times. Archived from the original on 31 July 2021. Retrieved 31 July 2021.
- ^ Champion, Paul (25 September 2007). "Professor in lese majeste row". Reuters. Archived from the original on 13 October 2007.
- ^ 2014 coup marks the highest number of lèse-majesté prisoners in Thai history Archived 1 May 2019 at the Wayback Machine. Prachatai.
- ^ "Thailand jails man for 35 years for insulting the monarchy on Facebook Archived 23 April 2018 at the Wayback Machine". The Independent. 10 June 2017.
- ^ "Thailand". freedomhouse.org. 5 January 2018. Archived from the original on 14 April 2018. Retrieved 3 May 2018.
- ^ a b "Thailand's top court tramples over the country's democracy". The Economist. 7 August 2024. Archived from the original on 16 August 2024. Retrieved 17 August 2024.
- ^ "Banning the opposition won't save Thailand's unpopular regime". The Economist. 7 August 2024. Archived from the original on 16 August 2024. Retrieved 17 August 2024.
- ^ "Thailand: Freedom in the World 2024 Report". freedomhouse.org. 29 February 2024. Archived from the original on 2 June 2024. Retrieved 29 February 2024.
- ^ "ประกาศกรมการปกครอง เรื่อง แจ้งข้อมูลทางการปกครอง" (PDF). กรมการปกครอง (in Thai). 13 March 2020. Archived from the original (PDF) on 20 May 2021. Retrieved 4 March 2018.
- ^ "LOCAL PERSONNEL ADMINISTRATION B.E. 2542 (1999)" (PDF). Department of Local Administration (DLA). Archived (PDF) from the original on 1 August 2019. Retrieved 11 December 2017.
- ^ Chivvis, Christopher S.; Marciel, Scot; Geaghan‑Breiner, Beatrix (26 October 2023). "Thailand in the Emerging World Order". Carnegie Endowment for International Peace. Retrieved 19 May 2024.
- ^ "The bamboo breaks: Thailand's diplomatic challenge". The Strategist. 10 September 2021. Archived from the original on 26 May 2022. Retrieved 27 April 2022.
- ^ Rakson, Katsamaporn (July–December 2018). "Investigating Thailand's self-perception in the regional context towards ASEAN". Veridian E-Journal, Silpakorn University. 11 (5): 568–578. Archived from the original on 10 August 2023. Retrieved 9 August 2023.
- ^ Chieocharnpraphan, Thosaphon (2015). "Strategic Partnership Between Australia and Thailand: A Case Study of East Timor". IAFOR Journal of Politics, Economics & Law. 2. doi:10.22492/ijpel.2.1.04. Archived from the original on 1 January 2024. Retrieved 1 January 2024.
- ^ OAS (1 August 2009). "OAS – Organization of American States: Democracy for peace, security, and development". OAS – Organization of American States. Archived from the original on 1 January 2024. Retrieved 1 January 2024.
- ^ "Thailand attends 2023 OSCE Asian Conference with aim to further promote Asia-Europe cooperation". กระทรวงการต่างประเทศ. Archived from the original on 1 January 2024. Retrieved 1 January 2024.
- ^ 'Thaksin to face charges over Burma telecom deal. ICT News, 2 August 2007
- ^ "Thai troops join Iraq force – Sep. 4, 2003". CNN. 4 September 2003. Archived from the original on 28 March 2024. Retrieved 20 December 2021.
- ^ "ภาพเก่าเล่าตำนาน : ทหารช่างไทย...ไปทำอะไร...ในอัฟกานิสถาน โดย พลเอก นิพัทธ์ ทองเล็ก". Matichon Online (in Thai). Archived from the original on 20 January 2024. Retrieved 1 January 2024.
- ^ The Telegraph, Troops from Thailand and Cambodia fight on border Archived 23 May 2010 at the Wayback Machine, 3 April 2009
- ^ Bloomberg, Thai, Cambodian Border Fighting Stops, Thailand Says Archived 14 October 2007 at the Wayback Machine
- ^ Prashanth Parameswaran. (2014). Thailand Turns to China Archived 27 April 2022 at the Wayback Machine. The Diplomat. Retrieved 3 January 2018.
- ^ "MPs warned of an economic colony as opposition zeroed in on Thailand's impaired relationship with China". Thai Examiner. 28 February 2020. Archived from the original on 19 October 2020. Retrieved 6 November 2020.
- ^ @Thavisin (7 October 2023). "In light of today's tragic incident..." (Tweet) – via Twitter.
- ^ "Israel-Palestine war: Thailand adopts neutral stance". nationthailand. 9 October 2023. Archived from the original on 16 October 2023. Retrieved 15 October 2023.
- ^ "คนไทยในอิสราเอลตายเพิ่ม 4 รวมเป็น 28 คน เร่งอพยพกลับไทย". Thai PBS (in Thai). Archived from the original on 16 October 2023. Retrieved 15 October 2023.
- ^ "Thailand Military Strength". Global Firepower. Archived from the original on 8 February 2015. Retrieved 15 December 2014.
- ^ Chapter 2 of the 2007 Constitution of Thailand
- ^ "_cf63a28daf.jpg (3194×2055)". กองบัญชาการกองทัพไทย (in Thai). Archived from the original on 1 January 2024. Retrieved 2 January 2024.
- ^ "World Bank Open Data". World Bank Open Data. Archived from the original on 26 May 2023. Retrieved 3 January 2024.
- ^ O'Sullivan, Michael; Subramanian, Krithika (17 October 2015). The End of Globalization or a more Multipolar World? (Report). Credit Suisse AG. Archived from the original on 15 February 2018. Retrieved 14 July 2017.
- ^ "Thailand Offers Persecuted Rohingya Little Hope". Human Rights Watch. 31 July 2019. Archived from the original on 9 June 2020. Retrieved 27 May 2020.
- ^ Battlefield Bangkok: The Royal Thai Army 2000–2014. Dean Wilson. 4 April 2015. ISBN 9781326046767. Archived from the original on 28 March 2024. Retrieved 1 July 2020.
- ^ Chapter 4 of the 2007 Constitution of Thailand
- ^ "Let's stop forcing boys to be soldiers" (Opinion). The Nation. 29 August 2018. Archived from the original on 28 August 2018. Retrieved 29 August 2018.
- ^ "Do away with conscription" (Opinion). Bangkok Post. 24 March 2018. Archived from the original on 28 March 2024. Retrieved 29 August 2018.
- ^ "Prawit denies servant for officer policy". Bangkok Post. 18 July 2018. Archived from the original on 28 March 2024. Retrieved 18 July 2018.
- ^ "Ex-private stands firm after being harassed online for criticising military". Pratchatai English. 3 November 2017. Archived from the original on 7 November 2017. Retrieved 4 November 2017.
- ^ "Conscripts aren't servants" (Opinion). Bangkok Post. 19 July 2018. Archived from the original on 28 March 2024. Retrieved 20 July 2018.
- ^ "Weeks after Korat massacre, Amnesty report describes conscript abuses". Bangkok Post. Reuters. 23 March 2020. Archived from the original on 28 March 2024. Retrieved 23 March 2020.
- ^ a b "Thailand's Deep State—The Military". Asia Sentinel. 14 November 2017. Archived from the original on 14 November 2017. Retrieved 15 November 2017.
- ^ "Thai Navy Accused of Involvement in Smuggling Rohingya Muslims". 19 July 2013. Archived from the original on 29 September 2020. Retrieved 27 May 2020.
- ^ ""บิ๊กติ๊ก"ตั้งลูก ติดยศทหาร อ้างให้งานทำ". Post Today. 16 April 2016. Archived from the original on 20 June 2019. Retrieved 29 August 2018.
- ^ "Thai Junta Fills Senate with Military, Police Officers". benarnews. Archived from the original on 26 September 2020. Retrieved 26 May 2020.
- ^ "2024 Global Peace Index" (PDF).
- ^ รายได้ประชาชาติของประเทศไทย พ.ศ. ๒๕๕๙ แบบปริมาณลูกโซ่ (in Thai). Office of the National Economic and Social Development Board. Archived from the original on 26 April 2018. Retrieved 23 April 2018.
- ^ a b c d ภาวะเศรษฐกิจไทยไตรมาสที่สี่ ทั้งปี 2560 และแนวโน้มปี 2561 (in Thai). Office of the National Economic and Social Development Board. 2018. Archived from the original on 27 April 2018. Retrieved 23 April 2018.
- ^ a b ภาวะสังคมไทยไตรมาสสี่และภาพรวม ปี 2560 (PDF) (in Thai). Office of the National Economic and Social Development Board. Archived from the original (PDF) on 26 April 2018. Retrieved 23 April 2018.
- ^ ข้อมูลหนี้สาธารณะคงค้าง (in Thai). Public Debt Management Office. Archived from the original on 26 April 2018. Retrieved 18 February 2018.
- ^ เสรีวรวิทย์กุล, ชนาภรณ์; รุ่งเจริญกิจกุล, ภูริชัย (July 2011). ฐานะทางการเงินของภาคครัวเรือนและผลของความมั่งคั่งต่อการบริโภค (PDF) (in Thai). Bank of Thailand. Archived from the original (PDF) on 10 October 2021. Retrieved 24 April 2018.
- ^ "GDP (PURCHASING POWER PARITY)". Central Intelligence Agency. Archived from the original on 30 December 2020. Retrieved 25 January 2019.
- ^ "NESDB: Thailand facing unemployment problem". Pattaya Mail. 25 November 2014. Archived from the original on 4 January 2015. Retrieved 4 January 2015.
- ^ "CThailand's GDP grows at fastest pace in 5 years in 2017". Nikkei Asian. Archived from the original on 6 March 2020. Retrieved 15 May 2020.
- ^ "Thailand raises public debt ceiling to fight COVID-19 outbreak". Reuters. 20 September 2021. Archived from the original on 21 April 2023. Retrieved 21 April 2023.
- ^ Allan, Juan (3 April 2024). "Thailand's economy lags behind peers with protracted recovery - Thailand Business News". www.thailand-business-news.com. Retrieved 24 July 2024.
- ^ Warr, Peter. "Thailand's economy remains beset by low productivity and slow growth". East Asia Forum. Retrieved 24 July 2024.
- ^ "Kiatnakin Phatra Research: Potential growth rate below 2%". Bangkok Post. Retrieved 24 July 2024.
- ^ a b Global Wealth Report 2016. Zurich: Credit Suisse AG. November 2016. Archived from the original on 15 May 2017. Retrieved 1 July 2017.
- ^ "Table 3: Inequality-adjusted Human Development Index". Human Development Report Office, United Nations Development Programme. Archived from the original on 29 January 2016. Retrieved 25 April 2018.
- ^ a b บทสรุปผู้บริหาร การสำรวจภาวะเศรษฐกิจและสังคมของครัวเรือน พ.ศ. 2560 (PDF) (in Thai). National Statistical Office. Archived from the original (PDF) on 26 April 2018. Retrieved 25 April 2018.
- ^ a b c รายงานการวิเคราะห์สถานการณ์ความยากจนและความเหลื่อมล้าในประเทศไทย ปี 2559 (PDF) (in Thai). Office of the National Economic and Social Development Board. 2016. Archived from the original (PDF) on 26 April 2018. Retrieved 24 April 2018.
- ^ "Profile of the Protestors: A Survey of Pro and Anti-Government Demonstrators in Bangkok on November 30, 2013" (PDF). Asia Foundation. December 2013. Archived from the original (PDF) on 26 April 2018. Retrieved 24 April 2018.
- ^ a b c d e พงศ์พิพัฒน์ บัญชานนท์ (18 June 2017). ยิ่งนานยิ่งถ่าง ช่องว่างทางรายได้ ปัญหาใหญ่ที่รอ คสช. แก้. BBC News ไทย (in Thai). BBC Thailand. Archived from the original on 14 May 2018. Retrieved 25 April 2018.
- ^ "แบงก์ชาติวิจัย "ไม่แข่ง-ยิ่งแพ้" เมื่อบริษัทใหญ่ 5% ครองรายได้ 85%". Prachachat (in Thai). Archived from the original on 21 April 2023. Retrieved 21 April 2023.
- ^ "Thai household debt surges, reaching a 16-year climax". Bangkok Post. Archived from the original on 28 March 2024. Retrieved 21 April 2023.
- ^ สสส. เผยสถานการณ์คนไร้บ้าน ทั่วประเทศกว่า 3 หมื่นคน. posttoday.com (in Thai). 16 June 2017. Archived from the original on 7 November 2018. Retrieved 25 April 2018.
- ^ "Asean Statistical Highlights 2023" (PDF). ASEAN Centre for Energy: 4. 2023.
- ^ Santivimolnat, Santan (18 August 2012). "2-million milestone edges nearer". Bangkok Post.
- ^ Languepin, Olivier (3 January 2013). "Thailand poised to Surpass Car Production target". Thailand Business News. Archived from the original on 15 January 2013. Retrieved 20 January 2013.
- ^ a b "Production Statistics". OICA (International Organization of Motor Vehicle Manufacturers). Archived from the original on 6 November 2013. Retrieved 26 November 2012.
- ^ a b Takahashi, Toru (27 November 2014). "Thailand's love affair with the pickup truck". Nikkei Asian Review. Archived from the original on 3 January 2015. Retrieved 4 January 2015.
- ^ Finlay, Steve (6 July 2012). "Pickup Trucks Reign in Thailand". Ward's. Archived from the original on 5 June 2019. Retrieved 5 June 2019.
- ^ "UNWTO World Tourism Barometer and Statistical Annex, December 2020 | World Tourism Organization". UNWTO World Tourism Barometer (English Version). 18 (7): 1–36. 18 December 2020. doi:10.18111/wtobarometereng.2020.18.1.7. S2CID 241989515.
- ^ "Government moves to head off tourist fears". Bangkok Post. 24 August 2015. Retrieved 24 August 2015.
- ^ Travel and Tourism, Economic Impact 2014: Thailand (PDF) (2014 ed.). London: World Travel & Tourism Council. 2014. Archived from the original (PDF) on 19 March 2015. Retrieved 10 March 2015.
- ^ Tourist Police in Thailand Archived 3 July 2008 at the Wayback Machine. Amazing-Thailand.com. Retrieved 16 September 2010.
- ^ "Medical Tourism in Thailand". MyMediTravel. Archived from the original on 18 February 2019. Retrieved 17 February 2019.
- ^ "Medical Tourism Report". WTTC. Archived from the original on 16 May 2020. Retrieved 15 May 2020.
- ^ Chokrungvaranont, Prayuth, Gennaro Selvaggi, Sirachai Jindarak, Apichai Angspatt, Pornthep Pungrasmi, Poonpismai Suwajo, and Preecha Tiewtranon. "The Development of Sex Reassignment Surgery in Thailand: A Social Perspective". The Scientific World Journal. Hindawi Publishing Corporation, 2014. Web. 23 March 2017.
- ^ Ocha, Witchayanee. "Transsexual emergence: gender variant identities in Thailand". Culture, Health & Sexuality14.5 (2012): 563–575. Web.
- ^ Thailand mulls legal prostitution. Archived 8 July 2011 at Wikiwix The Age, 26 November 2003
- ^ Martin, Lorna (25 January 2006). "Paradise Revealed". Taipei Times. Archived from the original on 2 December 2014. Retrieved 29 January 2015.
- ^ a b c d e Henri Leturque and Steve Wiggins 2010. Thailand's progress in agriculture: Transition and sustained productivity growth Archived 27 April 2011 at the Wayback Machine. London: Overseas Development Institute
- ^ International Grains Council. "Grain Market Report (GMR444)" Archived 2 July 2014 at the Wayback Machine, London, 14 May 2014. Retrieved 13 June 2014.
- ^ "CIA World Factbook – Greater Mekong Subregion". Central Intelligence Agency. Archived from the original on 26 March 2014. Retrieved 3 November 2011.
- ^ "Rice Around The World. Thailand". Irri.org. Archived from the original on 27 March 2008. Retrieved 25 April 2010.
- ^ a b "Country Trends". Global Footprint Network. Archived from the original on 8 August 2017. Retrieved 9 October 2019.
- ^ Lin, David; Hanscom, Laurel; Murthy, Adeline; Galli, Alessandro; Evans, Mikel; Neill, Evan; Mancini, Maria Serena; Martindill, Jon; Medouar, Fatime-Zahra; Huang, Shiyu; Wackernagel, Mathis (2018). "Ecological Footprint Accounting for Countries: Updates and Results of the National Footprint Accounts, 2012–2018". Resources. 7 (3): 58. doi:10.3390/resources7030058.
- ^ a b c Kongtip, Pornpimol; Nankongnab, Noppanun; Chaikittiporn, Chalermchai; Laohaudomchok, Wisanti; Woskie, Susan; Slatin, Craig (2015). "Informal Workers in Thailand: Occupational Health and Social Security Disparities". New Solutions. 25 (2): 189–211. doi:10.1177/1048291115586036. ISSN 1048-2911. PMC 5812466. PMID 25995374 – via National Library of Medicine.
- ^ Bales, Kevin (1999). Disposable People : New Slavery in the Global Economy. University of California Press. ISBN 9780520217973 – via Internet Archive.
- ^ a b Guille, Howard (2014). "Reforming Asian Labor Systems: Economic Tensions and Worker Dissent". Asian Studies Review. 39.
- ^ Warunsiri, Sasiwimon (2011). "The Role of Informal Sector in Thailand" (PDF). Research Institute for Policy Evaluation and Design. Archived from the original (PDF) on 11 April 2019. Retrieved 22 April 2018.
- ^ Coorlim, Leif (20 June 2014). "U.S. human trafficking report drops four nations to lowest tier". CNN. Archived from the original on 23 May 2022. Retrieved 23 May 2022.
- ^ Hodal, Kate; Kelly, Chris (10 June 2014). "Trafficked into slavery on Thai trawlers to catch food for prawns". The Guardian. ISSN 0261-3077. Retrieved 22 October 2024.
- ^ World Intellectual Property Organization (2024). Global Innovation Index 2024. Unlocking the Promise of Social Entrepreneurship (PDF). Geneva. p. 18. doi:10.34667/tind.50062. ISBN 978-92-805-3681-2. Retrieved 1 October 2024.
{{cite book}}:|website=ignored (help)CS1 maint: location missing publisher (link) - ^ "Research and development expenditure (% of GDP)". World Bank. Archived from the original on 19 May 2019. Retrieved 14 May 2020.
- ^ "Results of survey on R&D expenditure and manpower in 2019 announced". Office of National Higher Education Science Research and Innovation Policy Council |. Archived from the original on 25 October 2022. Retrieved 25 October 2022.
- ^ Thongkamkoon, Chaiwat (17 November 2017). "25601124-RaiwalDevOTP.pdf" (PDF). Office of Transport and Traffic Policy and Planning. Retrieved 3 January 2024.[permanent dead link]
- ^ Thongkamkoon, Chaiwat. "PowerPoint Presentation" (PDF). Thailand Board of Investment. Archived (PDF) from the original on 3 January 2024. Retrieved 3 January 2024.
- ^ Malaitham, Sathita (2013). "A Study Of Urban Rail Transit Development Effects In Bangkok Metropolitan Region" (PDF). Kyoto University.
- ^ เส้นทางปรับแผนรถไฟฟ้า. Mass Rapid Transit Master Plan in Bangkok Metropolitan Region website (in Thai). Office of Transport and Traffic Policy and Planning. Archived from the original on 2 January 2011. Retrieved 16 January 2012.
- ^ Janssen, Peter (23 January 2017). "Thailand's expanding state 'threatens future growth'". Nikkei Asian Review. Archived from the original on 24 November 2021. Retrieved 23 January 2017.
- ^ "Life and death on Thailand's lethal roads". BBC News. 19 January 2017. Archived from the original on 15 October 2019. Retrieved 17 June 2022.
- ^ Mahittirook, Amornrat (7 November 2016). "Public vans likely to offer 10% fare cut". Bangkok Post. Archived from the original on 28 March 2024. Retrieved 7 November 2016.
- ^ "The meter is ticking" (Opinion). Bangkok Post. 14 November 2018. Archived from the original on 6 June 2020. Retrieved 14 November 2018.
- ^ "Bangkok Suvarnabhumi Airport – FNM2024". Retrieved 19 May 2024.
- ^ a b "International Index of Energy Security Risk" (PDF). Institute for 21st Century Energy. 2013. Archived from the original (PDF) on 4 January 2015. Retrieved 14 September 2014.
- ^ a b "ASEAN Oil and Gas Updates 2023". ASEAN Centre for Energy. November 2023.
- ^ "Thailand energy report". Enerdata. Energy Supply. December 2023. Retrieved 22 October 2024.
- ^ "Alternative Energy Development Plan 2018-2037". Climate Change Laws of the World. 2020. Retrieved 22 October 2024.
- ^ "Shifting to alternative energy" (PDF). KPMG Phoomchai Audit Co. Ltd. January 2020.
- ^ "Thailand". World Health Organization. Retrieved 22 October 2024.
- ^ "The Population of Thailand from 1909 to 2000". National Statistical Office (Thailand). Archived from the original on 10 August 2023. Retrieved 10 August 2023.
- ^ "20230512163226_54316.pdf" (PDF). National Statistical Office. 2012. p. 12. Archived (PDF) from the original on 1 January 2024. Retrieved 2 January 2024.
- ^ "Average Household Size in Thailand". hub.arcgis.com. Archived from the original on 14 December 2023. Retrieved 14 December 2023.
- ^ Singh, Akanksha (3 June 2024). "Thailand's aging population hits consumption and economic growth - Thailand Business News". www.thailand-business-news.com. Retrieved 24 July 2024.
- ^ "Thailand", The World Factbook, Central Intelligence Agency, 15 October 2024, retrieved 22 October 2024
- ^ "Population total – Thailand". World Bank Group. Archived from the original on 13 October 2016. Retrieved 12 October 2016.
- ^ a b Ethnolinguistic Maps of Thailand (PDF) (in Thai). Office of the National Culture Commission. 2004. Archived from the original (PDF) on 9 October 2016. Retrieved 8 October 2016.
- ^ Luangthongkum, Theraphan (2007). "The Position of Non-Thai Languages in Thailand". Language, Nation and Development in Southeast Asia: 191.
- ^ Thailand: Burmese migrant children missing out on education. IRIN Asia. 15 June 2009. Archived 27 February 2012 at the Wayback Machine
- ^ McGeown, Kate (14 December 2006). "Hard lessons in expat paradise". BBC News. Archived from the original on 9 June 2011. Retrieved 1 March 2015.
- ^ "Speech to the Australian-Thai Chamber of Commerce". Australian Minister for Foreign Affairs and Trade. 3 July 2008. Archived from the original on 12 June 2019. Retrieved 12 November 2019.
- ^ a b c Lewis, M. Paul (2009). Ethnologue : languages of the world (16th ed.). Dallas, Texas: SIL International. pp. 529–533, 829–831. ISBN 978-1-55671-216-6.
- ^ Hartmann, John F. (1986), The spread of South Indic scripts in Southeast Asia, p. 8
- ^ "CERD/C/THA/1-3" (PDF). 5 October 2011. Archived from the original (PDF) on 9 October 2016. Retrieved 13 April 2024.
- ^ "Thailand | Ethnologue Free". Ethnologue (Free All). Archived from the original on 9 March 2023. Retrieved 12 April 2024.
- ^ "The Global Religious Landscape". Pew Research Center. December 2012. Archived from the original on 28 August 2014. Retrieved 5 November 2018.
- ^ Gerson, Ruth; Mallinger, Stephen Mark (2011). Jews in Thailand. Bangkok: River Books. ISBN 978-616-90895-0-6.
- ^ United States Bureau of Democracy, Human Rights and Labor. Thailand: International Religious Freedom Report 2007 Archived 10 November 2019 at the Wayback Machine. The article incorporates text from this source, which is in the public domain.
- ^ "2018 Report on International Religious Freedom: Thailand". US Department of State. Archived from the original on 8 December 2022. Retrieved 28 June 2021.
- ^ "No alcohol sales today – Makha Bucha Day". Thaiger. 8 February 2020. Archived from the original on 28 March 2024. Retrieved 31 July 2021.
- ^ "Education Reform at the Ministry of Education Thailand". elibrary.ksp.or.th. Archived from the original on 27 September 2023. Retrieved 8 October 2023.
- ^ "The Thailand Educational Reform Project" (PDF). backoffice.onec.go.th. Archived (PDF) from the original on 7 November 2023. Retrieved 8 October 2023.
- ^ "Thailand-Youth literacy rate". knoema. Archived from the original on 4 March 2021. Retrieved 13 May 2020.
- ^ Boonyatus, Jeerapa (28 June 2023). "Voices of students on school rules and uniforms". Thai PBS World.
- ^ "Thailand Provides 27,231 Schools With Internet". 11 March 2013. Archived from the original on 16 July 2014. Retrieved 30 January 2015.
- ^ "Covid hinders education again". Bangkok Post. Retrieved 10 August 2021.
- ^ "University Ranking". topuniversities. Archived from the original on 26 December 2020. Retrieved 13 May 2020.
- ^ Buasuwan, Prompilai (2018). "Rethinking Thai higher education for Thailand 4.0". Asian Education and Development Studies. 7 (2). emerald: 157–173. doi:10.1108/AEDS-07-2017-0072.
- ^ a b "Education in Thailand". WENR. 6 February 2018. Archived from the original on 19 September 2020. Retrieved 13 May 2020.
- ^ Charassangsomboon, Varissara (17 September 2018). "Exclusive: Thailand's plan to fight inequality in education". GovInsider. Retrieved 3 June 2024.
- ^ a b Draper, John (2012), "Revisiting English in Thailand", Asian EFL Journal, vol. 14, no. 4, pp. 9–38, ISSN 1738-1460, archived from the original on 12 March 2014
- ^ OECD (2013), Structural Policy Country Notes: Thailand (PDF), OECD, archived (PDF) from the original on 12 March 2014
- ^ Khaopa, Wannapa (12 December 2012). "Thai students drop in world maths and science study". The Nation. Archived from the original on 12 March 2014.
- ^ Draper, John (12 December 2011). "Solving Isaan's education problem". The Isaan Record. Archived from the original on 26 February 2013.
- ^ Draper, John (21 February 2014). "PISA Thailand regional breakdown shows inequalities between Bangkok and Upper North with the rest of Thailand". The Isaan Record. Archived from the original on 12 March 2014.
- ^ "English skills drop again". Bangkok Post. Retrieved 10 August 2021.
- ^ "สถิติอุดมศึกษา Higher Education Statistics 2558–2560" (PDF). Office of The higher Education Commission. Archived from the original (PDF) on 25 October 2021. Retrieved 13 May 2020.
- ^ "2019 Global Health Security Index". GHS INDEX. Archived from the original on 3 August 2020. Retrieved 15 May 2020.
- ^ "Search for JCI-Accredited Organizations". JCI. Archived from the original on 1 October 2020. Retrieved 15 May 2020.
- ^ Finch, Steve (7 January 2014). "Thailand top destination for medical tourists". Canadian Medical Association Journal. 186 (1): E1–E2. doi:10.1503/cmaj.109-4655. PMC 3883860. PMID 24246587.
- ^ "World malaria report 2023". World Health Organization. Design and layout by Claude Cardot, cover design by Lushomo: 22–23, 85. 20 November 2023. ISBN 978-92-4-008617-3.
{{cite journal}}: CS1 maint: others (link) - ^ Mingchay, Pichanon; Paitoonpong, Leilani; Kawkitinarong, Kamon; Ohata, Pirapon June; Suwanpimolkul, Gompol (20 August 2024). "Tuberculosis at a university hospital, Thailand: A surprising incidence of TB among a new generation of highly exposed health care workers who may be asymptomatic". PLOS ONE. 17 (8): e0273027. doi:10.1371/journal.pone.0273027. PMC 9401166. PMID 36001595.
- ^ "14,737 lives lost on Thai roads in 2022". nationthailand. 6 January 2023. Retrieved 20 October 2024.
- ^ Olam, Kocha; Goldschmidt, Debra (25 December 2018). "Thailand approves medical marijuana". CNN. Archived from the original on 26 December 2018. Retrieved 26 December 2018.
- ^ Wongworakul, Eve (21 December 2020). "History of Pad Thai as a Symbol of Nationalism in Thailand". arcgis.com. Archived from the original on 10 May 2024. Retrieved 9 May 2024.
- ^ Reynolds, E. Bruce (2004). "Phibun Songkhram and Thai Nationalism in the Fascist Era". European Journal of East Asian Studies. 3 (1). Brill: 99–134. doi:10.1163/1570061033004686. JSTOR 23615170. Archived from the original on 10 May 2024. Retrieved 9 May 2024.
- ^ Thepboriruk, Kanjana Hubik (August 2019). "Dear Thai Sisters: Propaganda, Fashion, and the Corporeal Nation under Phibunsongkhram" (PDF). Southeast Asian Studies. 8 (2): 233–258. Archived (PDF) from the original on 10 May 2024. Retrieved 9 May 2024.
- ^ Murray L Weidenbaum (1996). The Bamboo Network: How Expatriate Chinese Entrepreneurs are Creating a New Economic Superpower in Asia. Martin Kessler Books, Free Press. pp. 4–8. ISBN 978-0-684-82289-1.
- ^ "Thai Hospitality: Its Reputation and Culture | MMH in Asia Master Class in Bangkok". blogs.cornell.edu. Archived from the original on 1 January 2024. Retrieved 2 January 2024.
- ^ Smutkupt, Suriya (1976). A Descriptive Study of Thai Nonverbal Communication (Thesis). Portland State University. pp. 4, 31–32. doi:10.15760/etd.2584. Archived from the original on 18 February 2024. Retrieved 13 April 2024.
- ^ a b "Buddhist Arts of Thailand" (PDF). Archived (PDF) from the original on 11 December 2019. Retrieved 4 December 2019.
- ^ PCL, Post Publishing. "Wat Borommaniwat". Bangkok Post.
- ^ "โครงการจักทำองค์ความรู้ด้านการสำรวจสถาปัตยกรรมเพื่อการอนุรักษ์โบราณสถาน" (PDF). Fine Arts Department, Ministry of Culture(Thailand). Archived from the original (PDF) on 13 December 2019. Retrieved 13 December 2019.
- ^ "วัด" (PDF). Archived from the original (PDF) on 17 June 2020. Retrieved 13 December 2019.
- ^ Terwiel, Barend Jan (January 2007). Using Ockham's Razor with respect to the Ram Khamhaeng Controversy. "Breaking the Bonds" Hamburg 24–26 November 2006. Archived from the original on 30 May 2022. Retrieved 8 December 2017.
- ^ "Thai literature". Encyclopædia Britannica. Archived from the original on 19 October 2020. Retrieved 11 May 2020.
- ^ "การสร้างสรรค์ภาพประกอบศิลปะดิจิทัล: ภาพเย่เนี่เยอ บทแห่งชมกนในพระนิพนธ์เจ้าฟ้าธรรมธิเบศร (เจ้าฟ้ากุ้ง)" [Digital Art Illustration Creation: Ye Nie Ye, the Chapter of Chomkan in the writings of Prince Dharma Thibesra (Prince Kung)]. Journal of Humanities and Social Sciences Nakhon Pathom Rajabhat University (in Thai). 14 (2). 9 May 2024.
- ^ a b "Culture Overview : Literature and Performances". thaiembassy.
- ^ Scrima, Andrea (April 2019). "Duanwad Pimwana and Mui Poopoksakul with Andrea Scrima". The Brooklyn Rail. Retrieved 7 April 2019.
- ^ "Thai Traditional Dances – Dance costume Thailand". Thai to Siam. Archived from the original on 3 February 2020. Retrieved 12 May 2020.
- ^ Guffey, Ryan V.; Kaewkaen, Anothai (1 September 2017). "Historical Practices and Modern Interpretations: Understanding the Wai Khru Ceremony as a Thai Educational and Cultural Tradition". Journal of Educational Leadership in Action. 5 (1). Wai Khru: Guide to Rites and Symbolism. doi:10.62608/2164-1102.1049. ISSN 2164-1102.
- ^ Yang, Mi; Roongruang, Panya (30 May 2022). "The Mahori Music At Bangkok Thonburi University". Asia Pacific Journal of Religions and Cultures. 6 (1): 51.
- ^ "Expanding the Asean screen". Bangkok Post. Archived from the original on 10 February 2023. Retrieved 10 May 2020.
- ^ "Thai films get in on the action". The Hollywood Reporter. 3 April 2007. Archived from the original on 11 September 2020. Retrieved 10 May 2020.
- ^ "'Thai wave' in showbiz poised for big splash in China". NIKKEI Asian Review. Archived from the original on 3 June 2020. Retrieved 10 May 2020.
- ^ "From Singapore to Malaysia: Markets Leading the Expansion of Southeast Asian Cinema". The Hollywood Reporter. 16 May 2019. Archived from the original on 11 September 2020. Retrieved 10 May 2020.
- ^ Scheck, Frank (24 March 2008). ""Shutter" a bland horror remake". Reuters. Archived from the original on 15 September 2020. Retrieved 10 May 2020.
- ^ "Thailand's 'lakorn' soap operas come to PH". Philippine Daily Inquirer. 3 July 2018. Archived from the original on 3 June 2020. Retrieved 10 May 2020.
- ^ "The economic contribution of the film and television industries in Thailand" (PDF). Oxford Economics. Archived (PDF) from the original on 3 June 2020. Retrieved 10 May 2020.
- ^ "7 Rising Style Stars to Watch in 2020". VOGUE. 25 December 2019. Archived from the original on 3 June 2020. Retrieved 11 May 2020.
- ^ "Universal Music Group ดัน วิโอเลต วอเทียร์ เป็นศิลปินสากลเต็มตัว เปิดตัวแรง! จนเพลงจากอัลบั้ม Glitter and Smoke ติดท็อปชาร์ตถึง 12 ประเทศ | HITZ THAILAND". hitz.teroradio.com. 23 June 2020. Retrieved 10 September 2024.
- ^ "A Brief History on Pad Thai | Rice Bowl Deluxe". Rice Bowl Deluxe. 20 December 2021. Archived from the original on 1 January 2024. Retrieved 2 January 2024.
- ^ "Phad Thai Diplomacy: Get To Know The Best Thai Restaurants Outside Thailand". MICHELIN Guide. Archived from the original on 1 January 2024. Retrieved 1 January 2024.
- ^ "Thai Lemongrass – What is it and how is it used in Thai food?". 9 June 2021. Archived from the original on 1 January 2024. Retrieved 1 January 2024.
- ^ "Galangal as a Thai Food Ingredient". thaicookbook.tv. Archived from the original on 1 January 2024. Retrieved 1 January 2024.
- ^ "Turmeric as a Thai Food Ingredient". thaicookbook.tv. Archived from the original on 1 January 2024. Retrieved 1 January 2024.
- ^ "Coriander / Cilantro Leaves as a Thai Food Ingredient". thaicookbook.tv. Archived from the original on 1 January 2024. Retrieved 1 January 2024.
- ^ Tim Cheung (12 July 2017). "Your pick: World's 50 best foods". CNN. Archived from the original on 8 July 2017. Retrieved 5 May 2018.
- ^ "Mango Sticky Rice & Tub Tim Krob Listed in CNN's 50 Best Desserts Around The World". Buriram Times. 27 December 2018. Archived from the original on 1 January 2019. Retrieved 23 April 2022.
- ^ "World Rice Statistics Online Query Facility". International Rice Research Institute (IRRI). FAO. 2013. Archived from the original on 26 April 2016. Retrieved 30 January 2016.
- ^ Board, Jack (18 August 2019). "Food of the future? Five-star edible insects served up as Thailand gets creative with bug business". Channel News Asia (CNA). Archived from the original on 19 August 2019. Retrieved 19 August 2019.
- ^ "The 10 best street food cities in the world, per VirtualTourist.com, Frommer's". Daily News. New York. Agence France-Presse. Archived from the original on 1 December 2017. Retrieved 17 July 2022.
- ^ "The Hairy Bikers' Asian Adventure, Thailand – Bangkok and the Central Plains" (Video). BBC. Archived from the original on 4 December 2017. Retrieved 17 July 2022.
- ^ "Weights and measures in Thailand". Cockatoo.com. 17 December 1923. Archived from the original on 10 October 2017. Retrieved 25 April 2010.
- ^ "Top 10 Muay Thai Fighters You Should Know". muaythaicitizen. 5 July 2017. Archived from the original on 7 May 2020. Retrieved 11 May 2020.
- ^ Panahi, Majeed; Veroeveren, Pieter (12 June 2009). "Asian Nations Cup 1972". www.rsssf.org. Retrieved 20 October 2024.
- ^ "International Rugby Board – THAILAND". World Rugby. Archived from the original on 28 September 2011. Retrieved 25 April 2010.
- ^ The Nation Archived 25 April 2011 at the Wayback Machine, 19 July 2005
- ^ Nualkhair, Chawadee (10 July 2009). "Thailand woos foreign golfers with sun, sand traps". Reuters. Archived from the original on 15 July 2009. Retrieved 25 April 2010.
- ^ "Why to book with golf2thailand.com : Thailand Golf Courses Thailand Golf Packages". Golf2thailand.com. Archived from the original on 15 June 2006. Retrieved 25 April 2010.
- ^ "Chang Thailand Slammers – AirAsia ASEAN Basketball League". aseanbasketballleague.com. Archived from the original on 5 June 2012. Retrieved 2 June 2012.
- ^ "Thailand Basketball". best-basketball-tips.com. 18 May 2012. Archived from the original on 1 May 2012. Retrieved 2 June 2012.
- ^ "End of an era for Muay Thai at Lumpini". Bangkok Post. 6 February 2014. Archived from the original on 10 October 2017. Retrieved 6 February 2014.
- ^ "Rajamangala Stadium - Bangkok". The Stadium Guide. Retrieved 20 October 2024.
Further reading
- Chachavalpongpun, Pavin, ed. (2020). Routledge Handbook of Contemporary Thailand. Routledge. ISBN 9781138558410. OCLC 1110657073.
- Cooper, Robert George (2008). Culture Shock! Thailand: A Survival Guide to Customs and Etiquette. Marshall Cavendish Editions. ISBN 9789814828772. OCLC 1101343921.
- London, Ellen (2008). Thailand Condensed: 2000 Years of History & Culture. Marshall Cavendish Editions. ISBN 9789812615206.
- Lonely Planet's Best of Thailand. Lonely Planet guidebooks. Footscray, Vic.: Lonely Planet. 2020. OCLC 1312080896.
- Mishra, Patit Paban (2010). The History of Thailand. Greenwood. OCLC 548555562.
- Moore, Frank J., ed. (1974). Thailand: Its People, Its Society, Its Culture. HRAF Press. OCLC 722730.
- Wyatt, David K. (2003). Thailand: A Short History. Yale University Press. ISBN 9780300084757. OCLC 53392823.
- Zawacki, Benjamin (2021). Thailand: Shifting ground between the US and a rising China (2nd ed.). Bloomsbury. OCLC 1232148433.
External links
 Definitions from Wiktionary
Definitions from Wiktionary Media from Commons
Media from Commons News from Wikinews
News from Wikinews Quotations from Wikiquote
Quotations from Wikiquote Texts from Wikisource
Texts from Wikisource Textbooks from Wikibooks
Textbooks from Wikibooks Resources from Wikiversity
Resources from Wikiversity Travel information from Wikivoyage
Travel information from Wikivoyage
Government
- Thaigov.go.th – Government of Thailand
- Chief of State and Cabinet Members (archived 10 December 2008)
- Mfa.go.th – Ministry of Foreign Affairs
- Thailand Internet information – National Electronics and Computer Technology Center
- Ministry of Culture (archived 28 April 2015)
General information
- Thailand. The World Factbook. Central Intelligence Agency.
- Thailand entry in Library of Congress Country Studies. 1987
- Thailand from UCB Libraries GovPubs (archived 7 February 2009)
- Thailand from the BBC News
- Thailand Encyclopædia Britannica entry
 Wikimedia Atlas of Thailand
Wikimedia Atlas of Thailand- Longdo Map – Thailand maps in English and Thai
- Key Development Forecasts for Thailand from International Futures
- 2010 Thailand population census by Economic and Social statistics Bureau (archived 16 January 2013)
Travel
- Tourism Authority of Thailand – official tourism website
Other
- Thailand Country Fact Sheet from the Common Language Project (archived 31 July 2014)
- Southeast Asia Visions. "Browse the Southeast Asia Visions Collection". Cornell University Library.


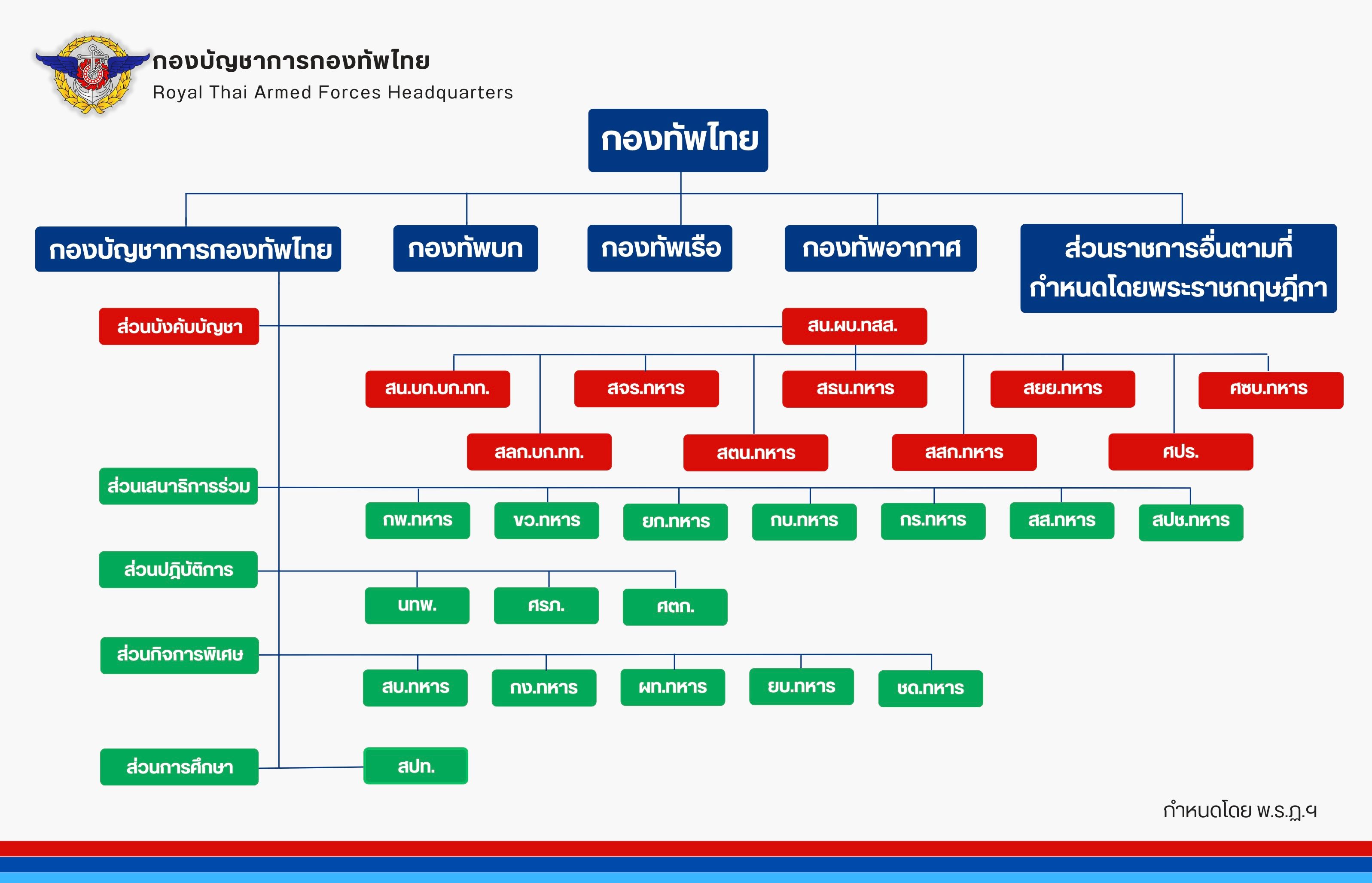{kind=link}
{kind=link}
| International | |
|---|---|
| National | |
| Geographic | |
| People | |
| Other | |
- Thailand
- 1932 establishments in Asia
- 1932 establishments in Siam
- 1932 establishments in Southeast Asia
- Countries in Asia
- Kingdoms
- Member states of ASEAN
- Member states of the United Nations
- Newly industrializing countries
- Observer states of the Organisation of Islamic Cooperation
- Southeast Asian countries
- States and territories established in 1932